
ABSTRACT
Histone modification map of H4 N-terminal tail residues in Saccharomyces cerevisiae reveals the prominence of lysine acetylation. Previous reports have indicated the importance of lysine acetylation in maintaining chromatin structure and function. H4K16, a residue with highly regulated acetylation dynamics has unique functions not overlapping with the other H4 N- terminal acetylable residues. The present work unravels the role of H4K16 acetylation in regulating expression of constitutive genes. H4K16 gets distinctly deacetylated over the coding region of constitutively expressed genes. Deacetylation of H4K16 reduces H3K9 acetylation at the cellular and gene level. Reduced H3K9 acetylation however did not negatively correlate with active gene transcription. Significantly, H4K16 deacetylation was found to be associated with hypoacetylated H4K12 throughout the locus of constitutive genes. H4K16 and K12 deacetylation is known to favour active transcription. Sas2, the HAT mutant showed similar patterns of hypoacetylated H3K9 and H4K12 at the active loci, clearly implying that the modifications were associated with deacetylation state of H4K16. Deacetylation of H4K16 was also concurrent with increased H3K56 acetylation in the promoter region and ORF of the constitutive genes. Combination of all these histone modifications significantly reduced H3 occupancy, increased promoter accessibility and enhanced RNAPII recruitment at the constitutively active loci. Consequently, we found that expression of active genes was higher in H4K16R mutant which mimic deacetylated state, but not in H4K16Q mimicking constitutive acetylation. To summarize, H4K16 deacetylation linked with H4K12 and H3K9 hypoacetylation along with H3K56 hyperacetylation generate a chromatin landscape that is conducive for transcription of constitutive genes.
Introduction
Functional fidelity of an eukaryotic genome is essentially maintained by regulated dynamism in chromatin. Post translational modifications of histones play a vital role in contributing to the flexibility of chromatin structure and function. In this context ‘histone code hypothesis’ has long proposed how an interplay of different histone modifications in various combinations forms the platform for chromatin changes during a nuclear process [Citation1–4]. The ‘access-repair-restore’ (ARR) model put forth decades back in context of DNA damage repair, holds true for any DNA metabolic process like transcription or replication [Citation5]. It emphasizes how timely modulation in chromatin structure makes DNA accessible or refractory to nuclear factors during a DNA metabolic process. This is a critical parameter that decides the proficiency of any DNA-dependent phenomenon like transcription, its initiation as well as regenerating the chromatin after completion of the process. An important modification that affects chromatin structure is acetylation of lysine residues that alters histone-DNA interactions and often facilitates recruitment of trans-factors that recognize acetylated lysines [Citation4,Citation6–10].
The histone H4 N-terminal tail has 4 acetylable lysine residues, namely, lysine 5 (H4K5), lysine 8 (H4K8), lysine 12 (H4K12) and lysine 16 (H4K16). Of these residues only H4K16 is known not to get methylated by virtue of its position [Citation11–13]. Lysine 16 of histone H4 N-terminal tail is a key player amongst the acetylable histone residues that actively contributes to the fidelity of chromatin structure [Citation9,Citation14,Citation15]. Consequently acetylation status of H4 K16 affects several nuclear functions such as gene silencing [Citation16–21], DNA repair [Citation22,Citation23] and transcription [Citation24–30]. In yeast, Hos2 mediated deacetylation of H4K16 is known to be required for active transcription [Citation29]. Earlier reports further indicate that deacetylation of H4K16 is a marker of human cancer and is correlated with reduced levels of DNA methylation [Citation31].
The chromatin environment of constitutively transcribed genes and the mode of their expression regulation is still not well understood. The current definitions of genes as constitutive or inducible do not include their regulatory state with respect to their position in chromatin domains. In Saccharomyces cerevisiae based on their activity and design of regulatory elements, genes are broadly classified into ‘growth genes’ or ‘housekeeping’ genes, that have constant levels of expression, encode ribosomal proteins, rRNA-processing enzymes, glycolytic enzymes and other factors required for rapid biomass production and ‘stress genes’ that have low levels of transcription under normal conditions but are induced to express at high levels under any form of stress [Citation32]. Transcription initiation in chromatin context is known to be initiated by formation of the pre-initiation complex (PIC), comprising of RNA Pol II and general transcription factors (TFIID/A/B/E/H) at the core promoter element aided by binding of activators to enhancers and recruitment of adaptor complexes SAGA and/or Mediator [Citation33–37]. Earlier classifications considered housekeeping genes to be lacking TATA boxes, having a well-defined NFR upstream of coding region and regulated by TFIID without aid of chromatin regulators like SAGA, while inducible genes contain TATA boxes and required the action of SAGA for access of TBP to promoters that remain occluded by nucleosomes [Citation32,Citation38,Citation39]. SAGA dominated genes were thought to be predominantly inducible and TFIID was considered to have a more housekeeping role. Inducible gene promoters were known to have more extensive chromatin regulation for successful transcription under inducible conditions [Citation32,Citation40]. Parallel studies however, also depict that over 99% of the measurable genome is positively regulated by the overlapping involvement of both TFIID and SAGA [Citation39,Citation41]. Additionally SAGA seems to get recruited to both TFIID and SAGA dominated promoters, irrespective of the presence of TATA box, signifying that SAGA acts as a general cofactor required for essentially all RNA polymerase II transcription [Citation42,Citation43]. The presence or absence of TATA box was rather found to affect the prevalence of histone modifications associated with transcription [Citation44]. The above studies have brought into perspective a different mode of classification of constitutive and inducible gene regulation that does not depend solely on TFIID or SAGA recruitment or presence or absence of TATA box in the gene promoters.
With this backdrop study on transcription of constitutive genes based on the combination of histone modifications that aids the process in a chromatin environment becomes an important avenue of investigation. Here we show that in Saccharomyces cerevisiae, deacetylation of H4K16 enhances transcription of constitutively expressed genes. Loss of acetylation in H4K16 was found to affect acetylation status of neighbouring lysine residues in H4 N-terminal tail and in the tail and core residues of histone H3 which enhanced promoter accessibility and RNA Pol II recruitment. This work thus sheds light on how deacetylation of H4K16 positively correlates with constitutive gene expression, due to an overall combinatorial effect of associated histone modifications that favour the process of transcription in the chromatin milieu of an eukaryotic cell.
Results
To understand the role of H4 K16 acetylation in transcription, studies were done with two histone mutant strains of yeast. One mutant strain H4 K16R has K16 of H4 mutated to arginine (R), to mimic the unacetylable state of lysine 16. The other mutant strain H4 K16Q has K16 of H4 mutated to glutamine (Q), to mimic constitutively acetylated state of lysine. Thereafter comparative studies were done in wild type and the two H4K16 mutants to understand the effect of H4K16 acetylation and associated chromatin modifications on transcription of constitutively expressed genes.
For the present study, six genes located on different chromosomes and having different physiological functions were chosen such that their level of expression would be constant throughout the cell cycle. PYK1 (YAL038 W), on chromosome I codes for pyruvate kinase which catalyses the final step of glycolysis i.e., conversion of phosphoenolpyruvate to pyruvate [Citation45–47]. TFC1 (YBR123 C), on chromosome II codes for the transcription factor Tau, one of the six subunits of the RNA polymerase III transcription initiation complex (TFIIIC) [Citation48,Citation49]. TAF10 (YDR167W), on chromosome IV codes for a TATA-binding-protein associated factor that is a subunit of the TFIID and the SAGA complex, both of which are required for the formation of the RNA polymerase II transcription initiation complex [Citation34–37,Citation50–52]. UBC6 (YER100W), on chromosome V codes for a ubiquitin conjugating enzyme (E2) present on the cytosolic membrane of the endoplasmic reticulum (ER), which functions in the ER associated proteasomal degradation (ERAD) pathway of misfolded proteins [Citation53,Citation54]. ACT1 (YFL039C), on chromosome VI encodes the single gene for the formation of yeast actin, which is a ubiquitous structural protein, conserved for various cytoskeletal functions like polarization of cell growth [Citation55,Citation56]. RPB2 (YOR151 C), on chromosome XV encodes the second largest subunit of the RNA polymerase II core enzyme, the absence or mutation of which leads to non- assembly of the core complex [Citation57].
Deacetylation of H4K16 promotes transcription of constitutively expressed genes
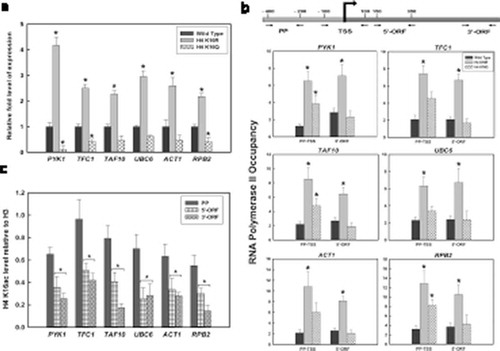
To check the effect of H4K16 acetylation on expression of constitutive genes, RT- qPCR based study was done. Comparative transcription analyses of six genes, PYK1, TFC1, TAF10, UBC6, ACT1 and RPB2, were done in wild type, H4K16R and H4K16Q cells. Results indicate that expression of these genes was significantly higher in H4K16R mutants, compared to wild type and H4K16Q ()). In H4K16Q cells, level of gene expression was rather distinctly reduced compared to wild type. Previous studies have shown that H4K16 acetylation in gene bodies is anticorrelated with transcription and presence of the mark towards 3’- ORF regions are transcription independent [Citation58]. These observations suggest that deacetylation of H4K16 promotes active gene transcription and acetylation of the residue has a negative correlation with the event. This was further correlated with RNAPII recruitment in the regions covering promoter and transcription start site (TSS) and 5’-ORF of the active genes, using specific antibody in a ChIP-based assay. It was very evident from our results that in the H4K16R mutant, both around the TSS and the 5’ end of coding region RNAPII recruitment was significantly enhanced compared to wild type ()). H4K16Q however had higher RNAPII recruitment than wild type in the promoter region but not in the 5’-ORF. This strengthens the earlier observation that transcription related events are favoured in absence of H4K16 acetylation. Thereafter, state of H4K16 acetylation was checked in wild type cells at the promoter proximal region and gene body of the above-mentioned loci using antibody against acetylated H4K16 followed by qPCR. Results indicated that H4K16 acetylation decreased significantly in the coding region of these genes compared to the promoter region ()). This clearly indicate that reduction in H4K16 acetylation was required for transcription of the constitutively expressing genes. Our observations find support with the previous report that Hos2-mediated deacetylation of H4K16 is required for transcription activation [Citation29]. Similar patterns of acetylated H4K16 at promoters followed by hypoacetylation at the gene body have also emerged from genome wide histone modification map of Drosophila [Citation59]. From the above results it is thus evident that deacetylation of H4K16 promotes transcription of constitutively expressed genes.
Figure 1. Constitutive Gene Expression in H4K16 mutants and the status of H4K16 acetylation during transcription. (a) RNA was isolated from wild type and H4 K16 mutant strains, followed by RT-qPCR to check expression of six constitutively active genes- PYK1, TFC1, TAF10, UBC6, ACT1 and RPB2. ChIP analyses with (b) RNAPII antibody to check recruitment at the promoter, TSS (PP-TSS) and 5’ end (5’-ORF) (c) anti-acetyl H4K16 antibody in wild type cells to check the level of acetylation at the promoter proximal region (PP), 5’ end (5’-ORF) and 3’end (3’-ORF) of the constitutively transcribing genes mentioned above. The H4K16 acetylation levels were normalized to ChIP data of the same regions with anti-H3 antibody and graphically plotted. Data represent the mean for three independent experiments with standard error of mean bars and asterisks indicate t-test significant P values < 0.05
Cellular levels of histone acetylation in wild-type and H4K16 mutants
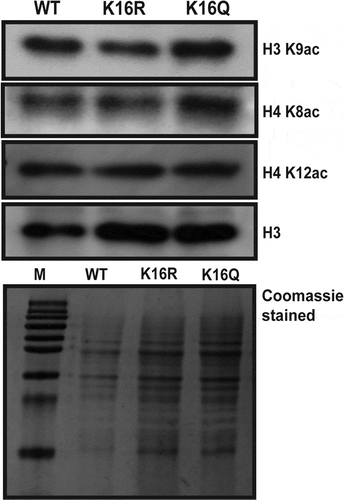
With an aim to understand the effect of H4K16 deacetylation on transcription of constitutive genes, cellular level acetylation of certain histone residues was next checked. In the H4 N-terminal tail, there are several acetylable histone lysine residues neighbouring H4K16. Among these, acetylation of H4K8 and K12 has been related to transcription. Acetylation of H4K8 in mammals has been shown to be required for binding of bromodomain containing proteins which function as co-activators and aid in the process of transcription activation [Citation60]. In actively transcribing genes Hos2-mediated deacetylation of H4K12 is known to occur predominantly at the coding region [Citation29]. To check H4K8 and K12 acetylation in wild-type and H4K16 mutants at the whole cell level, immunoblot assays were done with protein extracted from wild type, H4K16R and K16Q cells. As shown in , levels of H4K12 and K8 acetylation were similar in wild type and H4K16 mutants. When compared to the H3 levels in all the three strains, it was evident that H4 K8 and K12 acetylation levels had no significant difference between wild-type, H4K16R and K16Q mutants. The above observations indicate that constitutive acetylation or deacetylation of H4K16 do not differentially affect H4K8 and K12 acetylation at cellular level. Hereafter, acetylation level of another lysine residue H3K9 was compared in wild type and H4K16 mutant cells. H3K9 acetylation is known to be enriched in the promoter and around the TSS region of actively transcribing genes and promotes mammalian RNA Pol II towards transcription elongation [Citation43,Citation61]. When cellular levels of H3K9 acetylation was checked in wild-type and H4K16 mutants, it was observed that acetylation was lower in H4K16R compared to wild type and H4K16Q (). Such an observation indicated that deacetylation of H4K16 do not favour H3K9 acetylation. This observation is in consonance with an earlier report that has shown that Gcn5 the HAT responsible for acetylation of H3K9, prefers binding to acetylated H4K16 in vitro [Citation62]. Notably, even with lower H3K9 acetylation levels transcription of constitutive genes was not impeded but was rather higher in H4K16R cells, compared to wild type and H4K16Q.
Figure 2. Cellular level acetylation of histone H3 and H4 residues. Total protein was isolated from wild type (WT) and H4K16 mutant (K16R and K16Q) cells. Equal amount of protein from each strain was resolved in 15% SDS-PAGE and immunoblotted with antibodies against H3K9ac, H4K8ac, H4K12ac and H3, respectively. Membrane was incubated with horseradish peroxidase (HRP) – conjugated secondary antibody, treated with a chemiluminescent substrate and exposed to autoradiograph film for visualization
Deacetylated state of H4K16 affects H3K9 acetylation at gene level
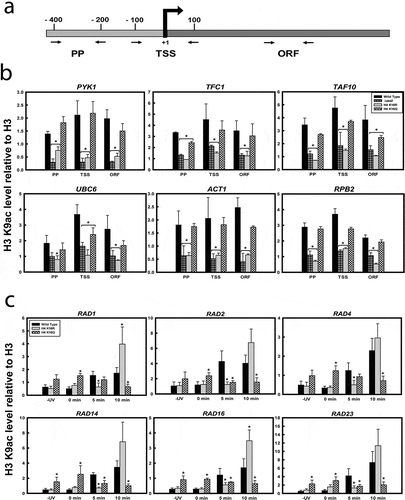
To further investigate, acetylation level of H3K9 was thereafter checked in the six constitutively expressing genes namely, PYK1, TFC1, TAF10, UBC6, ACT1 and RPB2, whose transcription was tested. For this, ChIP-qPCR was done using antibody against acetylated H3K9 and primers specific to promoter, TSS and ORF regions of the genes ()). As shown in ), in all the six genes tested, H3K9 acetylation levels were found to be distinctly reduced in H4K16R mutant, compared to wild type and H4K16Q. Lower H3K9 acetylation was found in the promoter and TSS region as well in the ORF of the genes. This observation is in continuity with the reduced cellular levels of H3K9 acetylation found in H4K16R cells, compared to wild type and H4K16Q (). To further confirm that reduced H3K9 acetylation was an effect of deacetylation of H4K16 (as H4K16R represents a constitutively deacetylated state of H4K16), Sas2- a HAT responsible for acetylation of H4K16 residue was deleted. When tested, H3K9 acetylation level in H4K16R and Sas2 mutant was found to be similarly reduced compared to wild type and H4K16Q cells ()). Interestingly, these observations further indicate that in constitutively active loci despite reduced H3K9 acetylation, relatively higher levels of gene expression were found in H4K16R compared to wild type and H4K16Q. To further understand the correlation between transcription and H3K9 acetylation, modification level was checked in six UV responsive genes, RAD1, RAD2, RAD4, RAD14, RAD16 and RAD23 under inducible conditions. For this, chromatin was isolated from all three kinds of cells treated with or without UV irradiations of 100 J/m2 and allowed to repair for 5 min or 10 min, respectively, followed by immunoprecipitation with anti-acetyl H3K9 antibody. Earlier we had shown that expression of these UV-inducible NER responsive genes was reduced in H4K16 mutants and the proteins showed a delayed recruitment at the NER site compared to wild type [Citation23]. Present results indicate that at the six inducible loci during early transcription induction (5 min), H3K9 acetylation levels were low in H4K16R cells, compared to wild type. The acetylation levels however increased at later time points post-UV-irradiation ()). In contrast, H3K9 acetylation levels did not vary in the H4K16Q cells pre- and post-induction. Notably, this was different from the pattern exhibited by wild type and H4K16R cells, both of which exhibited increased H3K9 acetylation following 5 min of transcription induction compared to the pre-induction level. Thus, in H4K16Q, H3K9 acetylation levels were higher than both wild type and H4K16R before transcription induction but reduced after UV irradiation. The exact reason for such constant H3K9 acetylation levels in H4K16Q cells before and after transcription induction needs to be further elucidated. A previous study [Citation63] has reported that H4K16R mutation causes upregulation of genes, especially of those that are regulated by chromatin. As expression of inducible genes is more intricately controlled by chromatin regulation an increase in H3K9 acetylation may be essential for the process. Hence unlike constitutive genes, for inducible gene expression a delayed but distinct increase in H3K9 acetylation was observed in the H4K16R cells. A constitutive hypoacetylation of H3K9 in inducible loci would possibly have graver effect on transcription of such genes, compared to what was observed for constitutive genes. In this context it has been previously shown that transcription initiation from inducible promoters requires recruitment of SAGA, the complex that includes Gcn5- although such recruitment may or may not depend on HAT activity of the complex [Citation64,Citation65]. To sum up, it thus becomes evident that when H4K16 is constitutively deacetylated lower H3K9 acetylation levels do not affect expression of constitutive genes. However, constitutive deacetylation of H4K16 cannot evade the requirement of H3K9 acetylation for expression of inducible genes.
Figure 3. H3K9 acetylation in wild-type and H4K16 mutants at the gene level. ChIP q- PCR analyses on chromatin isolated from wild type and the H4K16 mutant strains treated with or without UV radiation at 100 J/m2 followed by repair incubation for 5 or 10 min respectively, with anti-acetyl H3K9 antibody. (a) Schematic showing primers used for ChIP-qPCR, specific to promoter proximal region (PP), transcription start site (TSS) and mid ORF region (ORF) of genes. The levels of H3K9 acetylation in wild-type and H4K16 mutant cells were normalized to ChIP data of the same regions with anti-H3 antibody and graphically plotted to represent (b) constitutively expressing genes or (c) around the promoter and TSS region of UV-inducible genes. Data represent the mean for three independent experiments with standard error of mean bars and asterisks indicate t-test significant P values < 0.05
H4K16 acetylation state affects acetylation of other H4 N-terminal lysine residues
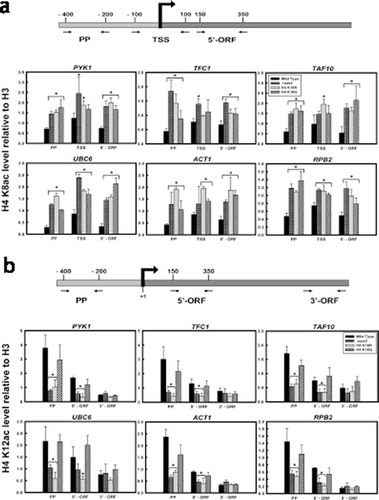
To further understand the correlation between H4K16 acetylation dynamics and chromatin regulation during transcription of constitutively expressing genes, the acetylation levels of H4 N-terminal lysine residues 8 and 12, was next checked at the gene level. ChIP- qPCR was done using antibodies against H4K8ac and H4K12ac, respectively followed by qPCR with primers spanning the promoter region and ORF of the six constitutively active genes mentioned above. H4K8 acetylation in both the H4K16 mutants and Sas2 was found to be higher compared to wild type, in the promoter region as well as over the gene body ()). Previously, it has been reported that H4K8 is the second-most redundantly acetylated H4 N-terminal lysine residue which gets acetylated in a transcription independent manner in yeast [Citation58,Citation63]. H4K8 acetylation in the mid and 3’-ORF regions of genes have been found to be either uncorrelated or anticorrelated with transcriptional activity in yeast genome [Citation27,Citation58]. Our observation of higher levels of H4K8 acetylation in both H4K16R and H4K16Q cells compared to wild type are in corroboration with the earlier reports of redundant H4K8 acetylation. It seems that irrespective of the deacetylated state of H4K16(R) which promotes transcription or the acetylated state of H4K16(Q) where transcription is hindered, H4K8 is apparently independently acetylated in a redundant manner. Hereafter, H4K12 acetylation level was checked at the constitutive loci. As shown in ), for all the genes tested, H4K16R mutants have reduced level of H4K12 acetylation compared to wild type and H4K16Q. The difference was especially evident in the promoter region, where H4K12 acetylation in the H4K16R cells was significantly lower than wild type and H4K16Q. While in wild type H4K12 acetylation gradually reduced over the gene body compared to promoter region, in H4K16R mutants H4K12 acetylation levels were low even at the promoter region and remained hypoacetylated in the 5’ ORF regions, as well. Sas2 mutant showed a similar pattern of reduced H4K12 acetylation in the promoter region and 5’ORF, compared to wild type. Unlike H4K16R and Sas2 mutants, H4K16Q cells did not show significantly reduced H4K12 acetylation level in the promoter or 5’ ORF regions, compared to wild type. As deacetylated H4K12 has been correlated with transcriptional activity [Citation29] increased H4K12 hypoacetylation in H4K16R than H4K16Q, especially at promoter region of constitutive genes may rationalize why gene expression is favoured in H4K16R compared to H4K16Q. Thus, a differential level of H4K12 acetylation is evident not only between wild type and H4K16R cells, but also between the two H4K16 mutants. As Hos2 mediated deacetylation of H4 K16 and K12 is essential for transcription activation, increased hypoacetylation of H4K12 in H4K16R mutants may provide a more conducive landscape for constitutive gene expression.
Figure 4. H4 K8 and K12 acetylation in wild type and H4K16 mutants at the gene level. ChIP-qPCR analyses with (a) anti-acetyl H4K8 antibody to check the level of acetylation at the promoter proximal region (PP), transcription start site (TSS) and 3’end of the ORFs (3’-ORF); (b) anti-acetyl H4K12 antibody to check the level of acetylation at the promoter proximal region (PP), 5’ end (5’-ORF) and 3’end (3’-ORF) of the ORFs of the constitutively transcribing genes mentioned above. The values were normalized to ChIP data of the same regions with anti-H3 antibody and graphically plotted to represent levels of H4 K8 and K12 acetylation in wild type and H4K16 mutant cells. Data represent the mean for three independent experiments with standard error of mean bars and asterisks indicate t-test significant P values < 0.05
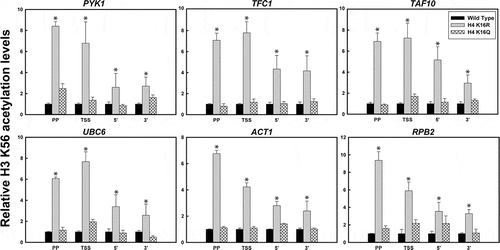
Deacetylation of H4K16 promotes acetylation of H3K56 residue
Acetylation of another histone H3 residue K56 is a hallmark for several DNA metabolic activities like replication, DNA damage repair and transcription that essentially involves nucleosome assembly and disassembly [Citation66–75]. This residue located at the entry-exit point of the DNA in a nucleosome is preferentially acetylated at the expressing loci and has been implicated as a marker of transcription activation [Citation71,Citation72,Citation74–76]. Next aim therefore was to understand whether H4K16 acetylation which contributes to chromatin structure and plays a role in metabolic functions like DNA repair and gene expression, has any correlation with H3K56 acetylation. ChIP-qPCR results clearly indicated significantly enhanced levels of H3K56 acetylation in the H4K16R mutant compared to wild type (). The higher levels of H3K56 acetylation in H4K16R was especially significant around the promoter and TSS region and was also prominent over the gene body. H4K16Q on the other hand was found to have H3K56 acetylation levels similar to wild type. From the above results it therefore becomes apparent that H4K16 deacetylation is associated with increased levels of H3K56 acetylation as observed in H4K16R cells. Whether activity of Rtt109, the primary HAT involved in H3K56 acetylation in yeast [Citation74,Citation75], is directly linked with H4K16 acetylation status needs to be pursued in future.
Figure 5. H3K56 acetylation pattern in H4K16 mutants. ChIP-qPCR analyses with anti- acetyl H3K56 antibody to check H3K56 acetylation pattern in transcriptionally active loci at the promoter proximal region (PP), transcription start site (TSS), 5’ end (5’-ORF) and 3’end (3’-ORF) of the ORFs of the constitutively active genes. The values were normalized to ChIP data of the same regions with anti-H3 antibody and graphically plotted to represent levels of H3K56 acetylation in H4K16 mutant cells relative to wild type. Data represent the mean for three independent experiments with standard error of mean bars and asterisks indicate t-test significant P values < 0.05
H3 occupancy and promoter accessibility in H4K16R cells
Earlier works have reported that H3K56 acetylation promotes high rate of histone exchange, especially prevalent near the nucleosome-free regions found at promoter sequences of both housekeeping and inducible genes and also facilitates RNA Pol II distribution along the expressing locus [Citation70,Citation71,Citation74,Citation75]. It has been further shown that absence of Rtt109 – the designated HAT, negatively affects eviction of histone H3, resulting in reduced RNAPII recruitment and decreased gene expression [Citation74,Citation75]. In this context, we hereafter checked histone occupancy in the active loci of wild type and H4K16 mutant cells. ChIP assay with anti-H3 antibody followed by qPCR at the six constitutively expressing loci indicated that H4K16R cells have distinctly reduced H3 occupancy at the promoter region, compared to wild type and H4K16Q ()). The difference was significant primarily in the promoter region and was not conspicuous over the gene body. This is consonance with previous reports that Rtt109 mediated acetylation of H3K56 is required for histone H3 eviction and RNAPII recruitment at the promoter region of active genes [Citation74,Citation75]. Overall, our results indicate that deacetylation of H4K16 residue is linked to increased acetylation of H3K56 which results in reduced H3 occupancy at the promoter regions of active genes.
Figure 6. Comparative histone occupancy and chromatin accessibility between wild type and H4K16 mutants. (a) ChIP-qPCR analyses with anti-H3 antibody for checking H3 occupancy at the promoter proximal region (PP), transcription start site (TSS), 5’ end (5’-ORF) and 3’end (3’-ORF) of the ORFs of transcriptionally active loci of wild type and H4K16 mutant cells. The values were calculated as percentage of input and graphically plotted to represent levels of H3 occupancy in H4K16 mutant cells relative to wild type. (b) DNase I digestion for different time points done with chromatin isolated from wild type and H4K16 mutant cells. Isolated DNA was subjected to qPCR with primers covering the promoter and TSS region. The amount of DNA amplified at each time point was normalized to the value of no digestion control. Normalized values from all the digestion time points was averaged and graphically plotted as normalized signal intensity for wild type and H4K16 mutants. Data represent the mean for three independent experiments with standard error of mean bars and asterisks indicate t-test significant P values < 0.05
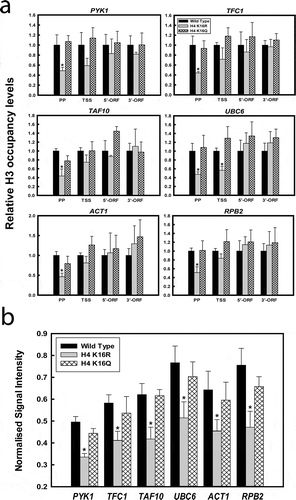
The above observations of increased H3K56 acetylation and reduced H3 occupancy in H4K16R cells was thereafter complemented with chromatin accessibility assay in the promoter region of these genes. For this, DNase I digestion was done with chromatin from wild type, H4 K16R and K16Q cells, for three different time points. Genomic DNA obtained following digestion reactions were subjected to qPCR using primers encompassing the promoter and TSS region of each of the genes. Logically, any differential chromatin accessibility in between the three strains will be reflected as a difference in DNase I accessibility in the loci. Consequently, higher DNase I accessibility will result in lower PCR signals, or the reverse. For all the strains, the PCR signals obtained from each time point for each gene was normalized against the signals obtained from no DNase I digestion control and averaged thereafter. Values thus obtained were graphically plotted as normalized signal intensity to represent inverse function of DNase I digestion. As shown in ), it was found that for all the genes tested the PCR signals obtained from H4K16R cells was noticeably lower than those obtained from wild-type and H4K16Q cells. It is thus conclusive that promoter region of the genes tested had higher DNase I digestion kinetics in H4K16R cells, compared to wild type and H4K16Q. Therefore, in H4K16R cells the promoter region of the constitutively active genes has increased chromatin accessibility, compared to wild type. Interestingly, in vitro experiments done earlier has shown that acetylation of H3K56 as found in H3K56Q cells, impedes nucleosome array oligomerization and is a probable mechanism for generating nucleosome free chromatin regions required for accessibility of trans factors during different DNA metabolic processes [Citation76].
Discussion
Acetylation of H4K16 has been previously described as a modification that acts as a switch to maintain heterochromatin-euchromatin structure and functions in S. cerevisiae [Citation16–20, Citation24–30]. In yeast, the modification dynamics of this particular residue is so critical that in addition to the common HAT (Esa1) and HDAC (Hos2) for the acetylable residues in the H4 N-terminal tail, H4 K16 has its own unique set of HAT (Sas2) and HDAC (Sir2) for maintaining its acetylation-deacetylation states [Citation18,Citation20,Citation25,Citation29,Citation77]. Acetylated state of H4 N- terminal tail is known to affect transcription efficiency and more specifically Sas2-mediated acetylation of H4K16 holds an inverse correlation with transcription elongation and RNA Pol II occupancy [Citation24,Citation25,Citation27,Citation58,Citation78]. Results from a whole genome transcriptional analysis done earlier concluded that of the four acetylable lysine residues present at the H4 N-terminal tail, only mutation in H4K16 produced specific effects on transcription, while mutation of others was partially redundant [Citation63]. In trying to further link H4K16 modification with transcription regulation, we deciphered how deacetylation state of H4K16 acts to promote expression of constitutive genes. The six genes tested were chosen from different yeast chromosomes with diverse functional roles. Additionally, four out of the six genes – UBC6, ACT1, TAF10 and TFC1 were without consensus TATA box sequences [TATA(A/T)A(A/T)(A/G)]; while two genes RPB2 and PYK1 had consensus TATA box sequences within 100 bp upstream of TSS [Citation38]. Our present observations are therefore of constitutively expressing genes from different chromosomal locations, with varied functions and irrespective of whether their promoters are TATA-less or TATA-containing. It was evident from our results that deacetylated state of H4K16, as found in the H4K16R mutant, facilitates several fold higher expressions of constitutively active genes. In wild type cells it was seen that acetylation of H4K16 significantly decreased over the gene body compared to promoter region, consistently for all the constitutive genes tested. Thus, normally a dynamism between acetylated and deacetylated states of H4K16 exists for the process of transcription. It has been shown earlier that Esa1-mediated acetylation of H4 N-terminal tail is essential for recruitment of TFIID and absence of such acetylation hampers transcription initiation leading to reduced expression of genes [Citation50,Citation65]. Precisely, such acetylation at the H4K16 residue at the promoter region of constitutively active genes is required for binding of Bdf1 protein through direct interaction with histones and subsequently aids in recruitment of TFIID [Citation79–81]. Since, TFIID is known to be required for expression of all mRNA in yeast [Citation41] and acetylated H4 tails works to anchor TFIID to the promoter during the initial stages of transcription activation [Citation82], the initial presence of acetylated H4K16 at the promoter region of the constitutive genes in wild type cells seems quite pertinent. Alongside, earlier experimental results have suggested that Sas2-mediated H4K16 acetylation poses an inhibitory effect on RNA Pol II travel over the gene body and consequently lower levels of H4K16 acetylation are found in genes that are frequently transcribed [Citation25]. Hypoacetylation of H4K16 has been found to be most prominent at −1 and +1 nucleosomes, but this is not restricted to these marks [Citation58]. Nucleosome mapping of PTMs on induction of stress has shown that H4K16 is acetylated on stress responsive genes at early time points but is eventually lost at later time points and deacetylated H4K16 is associated with the gene when transcriptional activity gets induced [Citation83]. As H4K16 acetylation at gene bodies has not been related to transcription [Citation58], rather deacetylation of H4K16 and H4K12 is shown to be required for transcription it is not surprising that in H4K16R where H4K12 acetylation is reduced expression of constitutive genes was higher compared to wild type. Notably, we also observed lower gene expression levels in H4K16Q cells compared to wild type, indicating that the lack of large-scale chromosome compaction observed in H4K16Q [Citation84] and removing H4K16 hypoacetylation by mimicking acetylated form might not confer a transcriptional advantage. Our observations thus underscore the importance of H4K16 hypoacetylation for effective transcription of constitutive genes in yeast.
Various combinations of acetylation patterns in H4-N terminal tail lysine residues, with certain extent of redundancy, has been correlated with several cellular functions in yeast, including transcription of different groups of genes [Citation27,Citation58,Citation63,Citation78,Citation85–87]. Our observation that elevated levels of H4K8 acetylation is present in the promoter and throughout the gene body of both H4K16 mutants in comparison to wild type, implied that acetylation of H4K8 and its role in transcription is independent of H4K16 acetylation status. On the contrary, the observation that the deacetylated state of H4K16 was distinctly associated with reduced levels of H4K12 acetylation was intriguing. Enrichment of hypoacetylated H4K12 in H4K16R and Sas2 mutant cells was especially prominent at the promoter region, compared to wild-type and H4K16Q cells. In wild type and H4K16Q, H4K12 hypoacetylation was enriched primarily in the coding region, but for H4K16R and Sas2 mutants hypoacetylated H4K12 was found throughout the active loci. These observations indicate a level of crosstalk between H4K16 and K12 residues in terms of acetylation and deacetylation during transcription. It is important to note in this context that NuA4 complex through its catalytic subunit Esa1 is responsible for acetylation of H4K12 along with H4K16, although abrogation of the HAT activity reduces H4K12 acetylation most strongly compared to all other substrates of Esa1 [Citation88]. Rpd3 and Hos2 the two HDACs responsible for deacetylation of H4 N-terminal tail residues, also have distinct roles. Rpd3 is known to be associated with actively expressed genes and is recruited throughout inducible loci under conditions of both activation and repression [Citation89–91]. Hos2 on the other hand, gets recruited only during transcription induction [Citation29] and thus seems to be more specifically required for the process of transcription. It has been previously shown that Hos2 mediates deacetylation of both H4K12 and H4K16 and this is required for activation of gene transcription. Deacetylation by Hos2 can occur in the coding and promoter region of actively transcribing genes and the transcription elongation factor Spt6 is known to specifically deposit deacetylated H4K16 on the ORFs during transcription [Citation29,Citation78]. Beyond S. cerevisiae, Hos2 in
U. maydis has also been shown to associate with the gene body, aid in transcription elongation and support maintenance of basal level gene expression [Citation92]. It is thus evident that Hos2-regulated deacetylation of H4K12 and K16 in transcriptionally active loci, is essential for gene expression. It is worth mentioning here that in wild type cells, acetylation of both H4K16 and K12 was found in the promoter region and hypoacetylation was observed more prominently at the coding sequences. While this may aid in transcription activation through recruitment of TFIID or SAGA [Citation41,Citation64,Citation65,Citation93] – hypoacetylation of both H4K16 and K12 residues at the promoter region of genes, as found in H4K16R mutant, must confer an additional benefit in the process. Lack of deacetylation due to deletion of Hos2 and Rpd3 has been shown to severely reduce RNAPII recruitment at the promoter region and consequently affect gene expression. It has been suggested that the deacetylation is required at a later stage of pre-initiation complex (PIC) formation and for multiple rounds of transcription initiation [Citation94]. This theory gains further support from our observation that RNAPII recruitment around the TSS and 5’-ORF regions is prominently enhanced in the H4K16R mutant, compared to wild type. Whether presence of deacetylated H4K16 and K12 at the promoter region of H4K16R mutant better facilitates PIC formation during transcription initiation compared to wild type, may be an interesting avenue to elucidate in future. Taken together, it is relevant to conclude from the present work that acetylation states of histones H4K16 and H4K12 crosstalk during transcription and the cumulative effect of deacetylation of these two residues promotes expression of constitutively active genes.
Acetylation of K9 in histone H3 has been linked to transcription through its association with active promoters and enhancers [Citation95]. Until recently direct functional role for H3K9ac during transcription was not known till it was shown that the acetylation was necessary to promote transcription elongation by recruitment of the super elongation complex (SEC) and for RNA Pol II pause release post initiation [Citation61]. It was further shown that lack of Gcn5 the HAT responsible for H3K9 acetylation, did not affect PIC formation [Citation61] indicating that H3K9 acetylation was required downstream to initiation. As per our observations, H3K9 acetylation was found to be significantly reduced in the promoter, around the TSS and in the ORF of constitutively expressed genes in H4K16R and Sas2 mutant cells. Notably previous in vitro studies have shown that, Gcn5 prefers acetylated H4K16 for binding and will not dock on the deacetylated residue [Citation62]. From all these studies it is thus clear that deacetylated state of H4K16 do not promote acetylation of H3K9, possibly due to inefficiency in binding of Gcn5. The fact that despite such low levels of H3K9 acetylation, expression of constitutively active genes was favoured in H4K16R mutant implies that H3K9 acetylation might not be absolutely indispensable for expression of such genes. This suggests that a bypass mechanism possibly exists for crucial steps such as PIC formation, RNA Pol II pause release or elongation when deacetylated H4K16 might not allow Gcn5 to acetylate H3K9. Interestingly, as mentioned above while NuA4 mediated H4 N-terminal tail acetylation in the promoter region of active genes is required for TFIID recruitment, in absence of functional NuA4, SAGA is involved in TBP recruitment to the promoter, irrespective of its HAT activity [Citation64,Citation65]. SEC recruitment has also been shown to be H4K5 acetylation dependent [Citation61] and H4K5ac is the most redundantly acetylated amongst all four lysine residues of the H4 N-terminal tail [Citation63]. It is therefore evident as to why lack of H4K16 acetylation at the promoter region, as found in the H4K16R cells, might not act as a stumbling block for constitutive gene expression. Thus, H4K16 acetylation-dependent and Gcn5-mediated H3K9 acetylation seems to be dispensable for transcription of constitutive genes. Our findings on enhanced constitutive gene expression in absence of H3K9 acetylation thus indicates a new avenue for analysing the requirement of histone acetylation during active transcription.
For inducible gene expression, however, H3K9ac seems to be necessary as even in H4K16R mutant acetylation was observed with progress in time of induction. Under such circumstances H3K9 acetylation may be mediated by one or more HAT other than Gcn5, whose recruitment is not dependent on H4K16 acetylation state. Recent findings show that regulated acetylation and deacetylation of H3K9 is required for different repair pathways and that H3K9 acetylation by different HATs is required for different functions of H3K9 in diverse chromatin processes [Citation23,Citation83,Citation96–99]. Rtt109 mediated acetylation of H3K9 has been found to be essential for tiding over the effects of genotoxic agents that block replication [Citation98,Citation100,Citation101]. Regulatory role of acetylated H3K9 during transcription is possibly mediated by HAT/coactivator p300, apart from the general H3 HAT Gcn5 [Citation99]. Thus, it is possible that other HATs capable of acetylating H3K9, whose recruitment and activity does not depend on H4K16 acetylation status, replace Gcn5 when H3K9 acetylation becomes absolutely necessary during transcription. This possibly explains why in H4K16R mutant delayed but distinct increase in H3K9 acetylation was observed for inducible gene expression.
Importantly, our present study shows a relation between acetylation of another histone H3 lysine residue with H4K16 acetylation state. Acetylation of the H3 core residue K56 was found to be significantly elevated when H4K16 was deacetylated. Enhanced H3K56 acetylation in H4K16 deacetylated cells was found throughout the loci, at the promoter region and throughout the gene body. Earlier it has been shown that during DNA damage response the effect of H3K56 hyperacetylation, can be supressed by reducing H4K16 acetylation levels [Citation102], thus indicating a probable crosstalk between these two histone modifications. Interestingly, in vitro studies have shown that Rtt109 when associated with Vps75 show HAT activity for H4 K12, but shows no such detectable activity for H4 residues as Rtt109-Asf1 complex [Citation100]. This indicates a possibility that Rtt109 as a HAT may have an affinity for H4 N-terminal lysine residues in general or specifically through its association with H4K12. This may work as a rate limiting step for H3K56 acetylation. When both H4K16 and K12 residues remain predominantly deacetylated, as seen in H4K16R cells, Rtt109 may be more readily available for H3K56 acetylation. This could possibly explain why enhanced levels of H3K56 acetylation was observed in constitutive loci of H4K16R cells, compared to wild type and H4K16Q. The direct correlation between these two modifications however remain to be established and needs further investigation. Furthermore, our observation that elevated levels of H3K56 acetylation in H4K16R cells was associated with significantly reduced H3 occupancy in the promoter region of all the constitutive tested genes is in agreement with the current understanding that H3K56 acetylation contributes during active transcription through its role in histone exchange and nucleosome disassembly [Citation25,Citation69–72,Citation74,Citation75,Citation83,Citation103–105]. While acetylated state of H3K56 has been distinctly correlated with nucleosome free region, it is well established that Rtt109-Asf1 mediated H3K56 acetylation although dispensable for PIC formation, is essential for histone eviction in transcriptionally active loci [Citation70,Citation74,Citation76,Citation106]. Therefore, deacetylated states of H4K16 and K12 along with highly acetylated H3K56 leads to reduced histone occupancy at the promoter region generating a chromatin landscape with increased accessibility. This is clearly evident from our DNase I accessibility assay results showing that chromatin around the promoter and TSS region of the constitutively active genes have higher accessibility in absence of H4K16 acetylation. The increased chromatin accessibility around the promoter region may have thus favoured transcription initiation while prevalence of H4K16 deacetylation in the coding region may have further promoted the process of transcription.
To summarize, our work here shows that deacetylation of H4K16 is a preferential histone modification for expression of constitutively active genes. Deacetylated state of H4K16 is part of a histone modification network that involves H4K12 deacetylation and H3K56 acetylation leading to a chromatin milieu with reduced H3 occupancy and increased DNA accessibility at the promoter region. The histone code so generated favours transcription of constitutive genes in yeast.
Materials and methods
Yeast strains
Saccharomyces cerevisiae strain WY121 was used as the parent strain to generate wild type, histone mutant and deletion mutant strains. Both copies of genomic histone H3-H4 are deleted in WY121 and one copy of H3-H4 is provided by plasmid pJL001 (CEN URA3 HHT2-HHF2) with URA3 as a counter-selectable marker. Plasmids pEMH7 (CEN TRP1 HHT2-HHF2), pEMH33 and pEMH35 with TRP1 selection marker, containing one copy of either wild type H3-H4 or point mutations of H4K16R or H4K16Q in the HHF2 gene coding for histone H4, were transformed into WY121 so that cells could now grow in synthetic SC−TRP-URA medium. Subsequently cells were grown in presence of 5’- FoA (5’-Fluoroorotic Acid) to shuffle out the URA3 containing pJL001 plasmid. These cells now containing only one plasmid served as the WT (pEMH7), K16R (pEMH33) and K16Q (pEMH35) strains. The WY121 strain and the pEMH plasmids were kind gifts from Dr. John Wyrick (Washington State University, USA) and Prof. J.D. Boeke (Johns Hopkins University School of Medicine, USA) respectively.
For generation of the Δsas2 strain, a PCR based homologous recombination-mediated gene deletion technique was used [Citation107]. Primers were designed for a selectable marker present on a shuttle vector, such that they had 5’ – overhangs with ~ 40 bp of sequence homology to regions flanking the SAS2 gene. The forward primer used was 5’- TCTAGTTGCTTTTTGTTTTCACTCGCAAAAAAAATGGCAAGATCTCAGCTGAAGC TTCGTACGC-3’ and the reverse primer used was 5’- TCCTGAAATACATATGCCATTAAGTTACATCCTGAATAGATTCCTAGCATAGGCC ACTAGTGGATCTG-3’. The 3’ end of the primer was specific for the KANMX4 module conferring resistance to antibiotic geneticin (G418), from the vector plasmid pFA6-KANMX4. The plasmid was a kind gift from Prof. Shubho Chaudhuri (Bose Institute, India). The PCR amplified linear DNA cassette containing selectable marker KANMX4 flanked by regions homologous to SAS2 was then transformed into WY121 cells using high efficiency yeast transformation protocol. The transformants were grown on G418 containing plates. Genomic DNA was isolated from colonies growing on G418 containing plates and subjected to PCR to confirm the deletion. PCR was done using forward primer corresponding to SAS2 specific region and the reverse primer in either the KANMX4 cassette or the SAS2 gene to serve as positive and negative controls. The genotype of the Δsas2 strain was further confirmed by Southern blotting.
Gene expression
Mid-log phase yeast cells with an O.D600 of ~ 0.7 (~ 1 × 107 cells/ml) were used to isolate RNA as previously described [Citation23]. Briefly, cell pellets were treated with a modified YLE buffer (1 M Sorbitol, 0.1 M EDTA pH 7.4, 0.1% β-mercaptoethanol, 50 U Zymolyase/107 cells), washed with Sorbitol wash buffer (1 M Sorbitol, 1 mM PMSF, 2 mM β-mercaptoethanol) and vortexed vigorously with Trizol® Reagent while keeping on ice. The mixture was then centrifuged at full speed for 10 minutes at 4°C to remove undissolved debris and the supernatant was used for chloroform extraction and subsequent precipitation of RNA pellets with isopropanol. 10 µg of RNA was DNaseI treated by Trizol®: chloroform extraction and isopropanol precipitation again. 5 µg of isolated RNA was used for reverse transcription using Revertaid RT (Thermo Scientific) as per manufacturer’s instructions. The cDNA formed was then serial diluted from 1/101 to 1/109 to serve as template for qPCR with primers for the six constitutive genes and the CT values obtained were used to plot a standard curve of CT values to number of amplicons (ng) present for each primer set. Subsequently, the undiluted cDNA was used for qPCR and the CT values of the expressed genes were then used on the standard curves to calculate the actual number of transcripts expressed for each gene using the standard formula: (amount of dsDNA in ng * Avogadro No.)/(Base pair size of dsDNA)*(330*2*D.F). The dilution factor (D.F) was taken as 109 and the amount of dsDNA present in ng was calculated as values of ‘x’ from the standard curve plot: y = mln(x) + c considering the CT values of the expressed genes as ‘y’. For each biological replicate (n = 3), triplicate qPCRs were repeated twice for each strain. The statistical significance of the difference in level of expression between wild type and mutant strains was tested using two-tailed independent student’s t-test and the results having a P value of < 0.05 were considered to be significant.
Chromatin immunoprecipitation
Log phase yeast cells with an OD600 of 0.9 ~ 1 (~ 1.5 × 107 cells/ml) were treated with 1% formaldehyde. For inducible genes, cells were treated or mock treated with 100 J/m2 UV light (254 nm), allowed to repair in the dark at 30°C for different time intervals (0, 5 and 10 mins) and then cross-linked with 1% formaldehyde. These cross-linked were harvested and washed with Sorbitol wash buffer (1 M sorbitol, 1 mM PMSF, 2 mM β-mercaptoethanol). Subsequent digestion with 50 units of Micrococcal Nuclease as mentioned previously [Citation21] was done, to generate 200–500 bp chromatin fragments and pre-cleared with pre-treated (50% slurry pre- absorbed with 0.1% BSA and 100 mg/ml of herring sperm DNA) Protein A-Sepharose beads at 4°C. 1% of the pre-cleared whole cell extract was kept as Input DNA and the rest was divided into equal aliquots and immunoprecipitated with 2–3 μg of antibody overnight at 4°C. The antibodies used were anti-Histone H4 Acetyl K16 (Cell Signalling Technology – E2B8W), anti-Histone H3 Acetyl K9 (abcam – ab10812), anti-Histone H4 acetyl K8 (abcam – ab15823), anti-Histone H4 acetyl K12 (abcam – ab46983), anti-Histone H3 acetyl K56 (abcam – ab195478), anti-RNA polymerase II (abcam – ab817) and anti-Histone H3 (BioBharati LifeScience – BB-AB0055). The immunocomplexes were precipitated with Protein A-Sepharose beads (pre- treated 50% slurry), washed with lysis buffer (50 mM HEPES–KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM PMSF), wash buffer 1 (Lysis buffer containing 500 mM NaCl), wash buffer 2 (10 mM Tris-Cl pH 8, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) and TE buffer, and then treated with RNase A. Immunocomplexes were then eluted from the beads using elution buffer (1% SDS, 0.1 M NaHCO3), crosslinks were reversed by incubation at 65°C and the de-cross-linked samples were treated with Proteinase K at 55°C. The DNA was isolated by phenol: chloroform extraction and ethanol precipitation and used for analysis by qPCR. Levels of histone modification were calculated using the formula 2(CT(Input) – CT (IP)) and were normalized by the levels of H3 present at each of the regions tested. H3 and RNA Polymerase II occupancy levels were calculated as a percentage of Input after extrapolating the 1% Input levels to 100%. For each ChIP, triplicate qPCRs were performed for three biological replicates (n = 3) and the data was plotted with standard error of mean bars. The statistical significance of the difference in levels of histone modifications between wild type and mutant strains was tested using two-tailed independent student’s t-test and the results having a P value of < 0.05 were considered to be significant.
Western blot
Whole cell lysates were prepared as per described protocol [Citation108], with brief modifications. Yeast cells were grown at 30°C in YPD to stationary phase with an O.D600 of 0.9–1.5 (~ 2–5 × 107 cells/ml), harvested, washed in ice cold 1X PBS with 1X protease inhibitor cocktail and pelleted. Cell pellets were lysed by rigorous vortexing with acid-washed glass beads (Sigma, 425–600 μm) in presence of ice-cold cell lysis buffer (50 mM HEPES pH 7.3, 200 mM NaCl, 1% Triton-X, 1X PIC, 1 mM PMSF). Samples were then centrifuged 15,000 g at 4°C for 15 minutes and the supernatant was used as whole cell extract (WCE). Protein concentration was estimated by Bradford Assay and 10 μg WCE was electrophoresed on 15% SDS-PAGE to blot samples on a PVDF membrane. Membrane was blocked with 5% NFD milk in TBS (25 mM Tris-Cl pH 7.5, 137 mM NaCl, 2.7 mM KCl) and incubated with primary antibody to the protein of interest in 1X TBS overnight at 4°C. Next day membrane was washed with 1X TBST (1X TBS, 0.1% Tween-20) and incubated with appropriate horseradish peroxidase (HRP) – conjugated secondary antibody for 1–3 hr at RT. Membrane was washed several times in excess volumes of 1X TBST. The membrane was thereafter incubated with a chemiluminescent substrate (SuperSignal® West Pico Chemiluminescent Substrate by Thermo Scientific), exposed to autoradiograph film and developed to visualize.
DNase I accessibility
Mid-log phase yeast cells with an O.D600 of ~ 0.7 (~ 1 × 107 cells/ml) were harvested and sphaeroplasted using YLE buffer (10 mg/ml Zymolyase 20 T in 1 M Sorbitol, 5 mM β- mercaptoethanol) by incubation at 22°C for 30 min. Hereafter previously described method [Citation109,Citation110] was followed with brief modifications. The sphaeroplasts were centrifuged at 500 g for 5 mins at 4°C and resuspended in ice-cold RSB buffer (10 mM Tris-Cl pH 7.4, 10 mM NaCl, 3 mM MgCl2) by flicking gently. Gentle lysis was done by addition of IGEPAL CA−360 at 0.05–0.1% final concentration followed by centrifugation at 500 g for 10 mins at 4°C. The white chromatin pellet thus obtained was resuspended in ice-cold RSB. Equal concentration of chromatin for each strain was determined based on Bradford Assay. Aliquots were taken and subjected to DNase I digestion using enzyme at a final concentration of 0.05 U/µl for increasing time points (0, 4, 8, 12 mins). Reactions were stopped by addition of 50 mM EDTA and heat inactivation at 65°C. DNA was isolated and subjected to qPCR with primers covering the promoter and TSS region. The amount of DNA amplified at each time point was normalized to the value of 0 min digestion control. Thereafter average of normalized values from all the time points was done. The average of the normalized values thus obtained was an inverse function of the accessibility to DNase I as lesser PCR signals indicated higher nuclease accessibility. Triplicate qPCRs were performed for three independent biological replicates (n = 3) and the data was plotted with standard error of mean bars. The statistical significance of the difference in accessibility to DNase I between wild type and mutant strains was tested using two-tailed independent student’s t-test and the results having a P value of < 0.05 were considered to be significant.
Authors’ contribution
RNC and AR designed the experiments; AR and PK performed the experiments; RNC, AR and PK analysed the results and wrote the paper.
Acknowledgements
We thank Prof. JD. Boeke (Johns Hopkins University School of Medicine, USA) for kind gift of the pEMH series of plasmids and our sincere acknowledgement to Prof. Michael J. Smerdon, Washington State University, USA for kind donation of the plasmids. We also sincerely thank Prof. John Wyrick (Washington State University, USA) for sharing the WY121 strain.
Disclosure statement
Contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the funding agency.
Additional information
Funding
References
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395.
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080.
- Shukla A, Chaurasia P, Bhaumik SR. Histone methylation and ubiquitination with their cross-talk and roles in gene expression and stability. Cell Mol Life Sci. 2009;66:1419–1433.
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45.
- Smerdon MJ. DNA repair and the role of chromatin structure. Curr Opin Cell Biol. 1991;3:422–428.
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643.
- Luger K, Mader AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260.
- Millar CB, Kurdistani SK, Grunstein M. Acetylation of yeast histone H4 lysine 16: a switch for protein interactions in heterochromatin and euchromatin. Cold Spring Harb Symp Quant Biol. 2004;69:193–200.
- Shogren-Knaak M, Peterson CL. Switching on chromatin: mechanistic role of histone H4-K16 acetylation. Cell Cycle. 2006;5:1361–1365.
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016.
- Fulton MD, Zhang J, He M, et al. Intricate effects of alpha- amino and lysine modifications on arginine methylation of the N-terminal tail of histone H4. Biochemistry. 2017;56:3539–3548.
- Green EM, Mas G, Young NL, et al. Methylation of H4 lysines 5, 8 and 12 by yeast Set5 calibrates chromatin stress responses. Nat Struct Mol Biol. 2012;19:361–363.
- Green EM, Morrison AJ, Gozani O. New marks on the block: set5 methylates H4 lysines 5, 8 and 12. Nucleus. 2012;3:335–339.
- Shogren-Knaak M, Ishii H, Sun JM, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847.
- Zhang R, Erler J, Langowski J. Histone acetylation regulates chromatin accessibility: role of H4K16 in inter-nucleosome interaction. Biophys J. 2017;112:450–459.
- Hecht A, Laroche T, Strahl-Bolsinger S, et al. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell. 1995;80:583–592.
- Johnson A, Li G, Sikorski TW, et al. Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol Cell. 2009;35:769–781.
- Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet. 2002;32:370–377.
- Oppikofer M, Kueng S, Martino F, et al. A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. Embo J. 2011;30:2610–2621.
- Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32:378–383.
- Kayne PS, Kim UJ, Han M, et al. Extremely conserved histone H4 N terminus is dispensable for growth but essential for repressing the silent mating loci in yeast. Cell. 1988;55:27–39.
- Bird AW, Yu DY, Pray-Grant MG, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–415.
- Ray A, Khan P, Nag Chaudhuri R. Regulated acetylation and deacetylation of H4 K16 is essential for efficient NER in saccharomyces cerevisiae. DNA Repair (Amst). 2018;72:39–55.
- Durrin LK, Mann RK, Kayne PS, et al. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell. 1991;65:1023–1031.
- Heise F, Chung HR, Weber JM, et al. Genome-wide H4 K16 acetylation by SAS-I is deposited independently of transcription and histone exchange. Nucleic Acids Res. 2012;40:65–74.
- Horikoshi N, Kumar P, Sharma GG, et al. Genome-wide distribution of histone H4 lysine 16 acetylation sites and their relationship to gene expression. Genome Integr. 2013;4:3.
- Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733.
- Taylor GC, Eskeland R, Hekimoglu-Balkan B, et al. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013;23:2053–2065.
- Wang A, Kurdistani SK, Grunstein M. Requirement of Hos2 histone deacetylase for gene activity in yeast. Science. 2002;298:1412–1414.
- Yu Y, Teng Y, Liu H, et al. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc Natl Acad Sci U S A. 2005;102:8650–8655.
- Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400.
- Rando OJ, Winston F. Chromatin and transcription in yeast. Genetics. 2012;190:351–387.
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719.
- Lee TI, Causton HC, Holstege FC, et al. Redundant roles for the TFIID and SAGA complexes in global transcription. Nature. 2000;405:701–704.
- Bhaumik SR, Green MR. SAGA is an essential in vivo target of the yeast acidic activator Gal4p. Genes Dev. 2001;15:1935–1945.
- Bhaumik SR, Green MR. Differential requirement of SAGA components for recruitment of TATA-box-binding protein to promoters in vivo. Mol Cell Biol. 2002;22:7365–7371.
- Li XY, Bhaumik SR, Green MR. Distinct classes of yeast promoters revealed by differential TAF recruitment. Science. 2000;288:1242–1244.
- Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709.
- Huisinga KL, Pugh BF. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell. 2004;13:573–585.
- Nagai S, Davis RE, Mattei PJ, et al. Chromatin potentiates transcription. Proc Natl Acad Sci U S A. 2017;114:1536–1541.
- Warfield L, Ramachandran S, Baptista T, et al. Transcription of nearly all yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Mol Cell. 2017;68:118–129 e115.
- Baptista T, Grunberg S, Minoungou N, et al. SAGA is a general cofactor for RNA polymerase II transcription. Mol Cell. 2017;68:130–143 e135.
- Bonnet J, Wang CY, Baptista T, et al. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 2014;28:1999–2012.
- Natsume-Kitatani Y, Mamitsuka H. Classification of promoters based on the combination of core promoter elements exhibits different histone modification patterns. PloS One. 2016;11:e0151917.
- Burke RL, Tekamp-Olson P, Najarian R. The isolation, characterization, and sequence of the pyruvate kinase gene of Saccharomyces cerevisiae. J Biol Chem. 1983;258:2193–2201.
- Fraenkel DG. The top genes: on the distance from transcript to function in yeast glycolysis. Curr Opin Microbiol. 2003;6:198–201.
- Pearce AK, Crimmins K, Groussac E, et al. Pyruvate kinase (Pyk1) levels influence both the rate and direction of carbon flux in yeast under fermentative conditions. Microbiology. 2001;147:391–401.
- Parsons MC, Weil PA. Cloning of TFC1, the Saccharomyces cerevisiae gene encoding the 95-kDa subunit of transcription factor TFIIIC. J Biol Chem. 1992;267:2894–2901.
- Swanson RN, Conesa C, Lefebvre O, et al. Isolation of TFC1, a gene encoding one of two DNA-binding subunits of yeast transcription factor tau (TFIIIC). Proc Natl Acad Sci U S A. 1991;88:4887–4891.
- Uprety B, Lahudkar S, Malik S, et al. The 19S proteasome subcomplex promotes the targeting of NuA4 HAT to the promoters of ribosomal protein genes to facilitate the recruitment of TFIID for transcriptional initiation in vivo. Nucleic Acids Res. 2012;40:1969–1983.
- Klebanow ER, Poon D, Zhou S, et al. Isolation and characterization of TAF25, an essential yeast gene that encodes an RNA polymerase II-specific TATA-binding protein-associated factor. J Biol Chem. 1996;271:13706–13715.
- Tora L. A unified nomenclature for TATA box binding protein (TBP)-associated factors (TAFs) involved in RNA polymerase II transcription. Genes Dev. 2002;16:673–675.
- Gilon T, Chomsky O, Kulka RG. Degradation signals recognized by the Ubc6p- Ubc7p ubiquitin-conjugating enzyme pair. Mol Cell Biol. 2000;20:7214–7219.
- Sommer T, Jentsch S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 1993;365:176–179.
- Pruyne D, Bretscher A. Polarization of cell growth in yeast. J Cell Sci. 2000;113(Pt 4):571–585.
- Pruyne D, Bretscher A. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J Cell Sci. 2000;113(Pt 3):365–375.
- Archambault J, Friesen JD. Genetics of eukaryotic RNA polymerases I, II, and III. Microbiol Rev. 1993;57:703–724.
- Liu CL, Kaplan T, Kim M, et al. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328.
- Bell O, Wirbelauer C, Hild M, et al. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. Embo J. 2007;26:4974–4984.
- Gaucher J, Boussouar F, Montellier E, et al. Bromodomain-dependent stage-specific male genome programming by Brdt. Embo J. 2012;31:3809–3820.
- Gates LA, Shi J, Rohira AD, et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J Biol Chem. 2017;292:14456–14472.
- Owen DJ, Ornaghi P, Yang JC, et al. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. Embo J. 2000;19:6141–6149.
- Dion MF, Altschuler SJ, Wu LF, et al. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A. 2005;102:5501–5506.
- Shukla A, Bajwa P, Bhaumik SR. SAGA-associated Sgf73p facilitates formation of the preinitiation complex assembly at the promoters either in a HAT-dependent or independent manner in vivo. Nucleic Acids Res. 2006;34:6225–6232.
- Uprety B, Sen R, Bhaumik SR. Eaf1p is required for recruitment of NuA4 in targeting TFIID to the promoters of the ribosomal protein genes for transcriptional initiation in vivo. Mol Cell Biol. 2015;35:2947–2964.
- Bernier M, Luo Y, Nwokelo KC, et al. Linker histone H1 and H3K56 acetylation are antagonistic regulators of nucleosome dynamics. Nat Commun. 2015;6:10152.
- Hyland EM, Cosgrove MS, Molina H, et al. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:10060–10070.
- Maas NL, Miller KM, DeFazio LG, et al. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–119.
- Masumoto H, Hawke D, Kobayashi R, et al. A role for cell-cycle- regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298.
- Rufiange A, Jacques PE, Bhat W, et al. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405.
- Topal S, Vasseur P, Radman-Livaja M, et al. Distinct transcriptional roles for histone H3-K56 acetylation during the cell cycle in Yeast. Nat Commun. 2019;10:4372.
- Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385.
- Yang J, Zhang X, Feng J, et al. The histone chaperone FACT contributes to DNA replication-coupled nucleosome assembly. Cell Rep. 2016;14:1128–1141.
- Schneider J, Bajwa P, Johnson FC, et al. Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem. 2006;281:37270–37274.
- Durairaj G, Chaurasia P, Lahudkar S, et al. Regulation of chromatin assembly/disassembly by Rtt109p, a histone H3 Lys56-specific acetyltransferase, in vivo. J Biol Chem. 2010;285:30472–30479.
- Watanabe S, Resch M, Lilyestrom W, et al. Structural characterization of H3K56Q nucleosomes and nucleosomal arrays. Biochim Biophys Acta. 2010;1799:480–486.
- Allard S, Utley RT, Savard J, et al. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. Embo J. 1999;18:5108–5119.
- Reiter C, Heise F, Chung HR, et al. A link between Sas2- mediated H4 K16 acetylation, chromatin assembly in S-phase by CAF-I and Asf1, and nucleosome assembly by Spt6 during transcription. FEMS Yeast Res. 2015;15:fov073.
- Jacobson RH, Ladurner AG, King DS, et al. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425.
- Matangkasombut O, Buratowski S. Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell. 2003;11:353–363.
- Pamblanco M, Poveda A, Sendra R, et al. Bromodomain factor 1 (Bdf1) protein interacts with histones. FEBS Lett. 2001;496:31–35.
- Martinez-Campa C, Politis P, Moreau JL, et al. Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail and the bromodomain factor Bdf1. Mol Cell. 2004;15:69–81.
- Weiner A, Hsieh TH, Appleboim A, et al. High-resolution chromatin dynamics during a yeast stress response. Mol Cell. 2015;58:371–386.
- Hsieh TH, Weiner A, Lajoie B, et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 2015;162:108–119.
- Goudarzi A, Zhang D, Huang H, et al. Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol Cell. 2016;62:169–180.
- Ma XJ, Wu J, Altheim BA, et al. Deposition-related sites K5/K12 in histone H4 are not required for nucleosome deposition in yeast. Proc Natl Acad Sci U S A. 1998;95:6693–6698.
- Ruan K, Yamamoto TG, Asakawa H, et al. Histone H4 acetylation required for chromatin decompaction during DNA replication. Sci Rep. 2015;5:12720.
- Chang CS, Pillus L. Collaboration between the essential Esa1 acetyltransferase and the Rpd3 deacetylase is mediated by H4K12 histone acetylation in Saccharomyces cerevisiae. Genetics. 2009;183:149–160.
- De Nadal E, Zapater M, Alepuz PM, et al. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature. 2004;427:370–374.
- Kurdistani SK, Robyr D, Tavazoie S, et al. Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat Genet. 2002;31:248–254.
- Robert F, Pokholok DK, Hannett NM, et al. Global position and recruitment of HATs and HDACs in the yeast genome. Mol Cell. 2004;16:199–209.
- Elias-Villalobos A, Helmlinger D, Ibeas JI. Histone deacetylases: revealing the molecular base of dimorphism in pathogenic fungi. Microb Cell. 2015;2:491–493.
- Ferdoush J, Sen R, Kaja A, et al. Two distinct regulatory mechanisms of transcriptional initiation in response to nutrient signaling. Genetics. 2018;208:191–205.
- Sharma VM, Tomar RS, Dempsey AE, et al. Histone deacetylases RPD3 and HOS2 regulate the transcriptional activation of DNA damage-inducible genes. Mol Cell Biol. 2007;27:3199–3210.
- Karmodiya K, Krebs AR, Oulad-Abdelghani M, et al. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012;13:424.
- Guo R, Chen J, Mitchell DL, et al. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res. 2011;39:1390–1397.
- Meyer B, Fabbrizi MR, Raj S, et al. Histone H3 lysine 9 acetylation obstructs ATM activation and promotes ionizing radiation sensitivity in normal stem cells. Stem Cell Reports. 2016;7:1013–1022.
- Chen Y, Zhang Y, Ye H, et al. Structural basis for the acetylation of histone H3K9 and H3K27 mediated by the histone chaperone Vps75 in Pneumocystis carinii. Signal Transduct Target Ther. 2019;4:14.
- Zhou W, Jiang D, Tian J, et al. Acetylation of H3K4, H3K9, and H3K27 mediated by p300 regulates the expression of GATA4 in cardiocytes. Genes Dis. 2019;6:318–325.
- Abshiru N, Ippersiel K, Tang Y, et al. Chaperone-mediated acetylation of histones by Rtt109 identified by quantitative proteomics. J Proteomics. 2013;81:80–90.
- Cote JM, Kuo YM, Henry RA, et al. Two factor authentication: Asf1 mediates crosstalk between H3 K14 and K56 acetylation. Nucleic Acids Res. 2019;47:7380–7391.
- Simoneau A, Delgoshaie N, Celic I, et al. Interplay between histone H3 lysine 56 deacetylation and chromatin modifiers in response to DNA damage. Genetics. 2015;200:185–205.
- Tan Y, Xue Y, Song C, et al. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 2013;110:11493–11498.
- Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc Natl Acad Sci U S A. 2008;105:9000–9005.
- Xie W, Song C, Young NL, et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33:417–427.
- Kaplan T, Liu CL, Erkmann JA, et al. Cell cycle- and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet. 2008;4:e1000270.
- Gardner JM, Jaspersen SL. Manipulating the yeast genome: deletion, mutation, and tagging by PCR. Methods Mol Biol. 2014;1205:45–78.
- Szymanski EP, Kerscher O. Budding yeast protein extraction and purification for the study of function, interactions, and post-translational modifications. J Vis Exp. 2013;80:e50921.
- Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc. 2010;2010:pdb prot5384.
- Zhong J, Luo K, Winter PS, et al. Mapping nucleosome positions using DNase-seq. Genome Res. 2016;26:351–364.