
ABSTRACT
Cigarette smoking impacts DNA methylation, but the investigation of sex-specific features of lung tissue DNA methylation in smokers has been limited. Women appear more susceptible to cigarette smoke, and often develop more severe lung disease at an earlier age with less smoke exposure. We aimed to analyse whether there are sex differences in DNA methylation in lung tissue and whether these DNA methylation marks interact with smoking. We collected lung tissue samples from former smokers who underwent lung tissue resection. One hundred thirty samples from white subjects were included for this analysis. Regression models for sex as a predictor of methylation were adjusted for age, presence of COPD, smoking variables and technical batch variables revealed 710 associated sites. 294 sites demonstrated robust sex-specific methylation associations in foetal lung tissue. Pathway analysis identified 6 nominally significant pathways including the mitophagy pathway. Three CpG sites demonstrated a suggested interaction between sex and pack-years of smoking: GPR132, ANKRD44 and C19orf60. All of them were nominally significant in both male- and female-specific models, and the effect estimates were in opposite directions for male and female; GPR132 demonstrated significant association between DNA methylation and gene expression in lung tissue (P < 0.05). Sex-specific associations with DNA methylation in lung tissue are wide-spread and may reveal genes and pathways relevant to sex differences for lung damaging effects of cigarette smoking.
Background
Cigarette smoking impacts diverse health problems including respiratory disease [Citation1,Citation2], cardiovascular disease [Citation3,Citation4], various kinds of cancer [Citation5,Citation6] and premature death [Citation7,Citation8]. Cigarette smoke contains a mixture of over 6000 chemicals, and many of them reach the lower respiratory tract to cause inflammation [Citation9]. Understanding the underlying molecular mechanisms of cigarette smoke toxicity is still a pressing scientific focus for research. Since the minority of smokers develop lung disease, and chronic obstructive pulmonary disease (COPD) demonstrates differences by sex [Citation10], modelling interactions between genetic and environmental influences on COPD are important. DNA methylation is an epigenetic process that regulates gene expression and genome stability [Citation11]. DNA methylation can be modified by both genetic variation and environmental exposures, which may impact the development of COPD and COPD-related phenotypes [Citation12]. Several studies have explored the associations between smoking and DNA methylation [Citation11,Citation13–21]. However, little is known about the sex-specific molecular effects of smoking on DNA methylation. Most DNA methylation studies of COPD were performed using peripheral blood cells [Citation13–21]; studies evaluating the sex-specific effects of smoking in lung tissue are scarce [Citation22]. Results from whole blood may be influenced by altered individual immune cell types caused by smoking. Furthermore, gene expression is, at least partially, a tissue-specific phenomenon [Citation23]. Though the prevalence of smoking is lower in women than in men, some studies have suggested that women are more susceptible to the lung damaging effect of smoking and often experience more severe lung disease despite lower or equal smoking exposure to men [Citation24–30]. Our aim was to identify sex-specific DNA methylation marks in adult lung tissue and to investigate sex by smoking interactions that may be relevant to understanding the sex-specific impact of smoking on lung disease risk. DNA methylation associations with sex in adult lung tissue were replicated in foetal lung tissue and also compared with published sex-specific observations in cord blood to evaluate lung specificity of overlapping foetal-adult lung tissue associations [Citation31]. Sex-specific associations with DNA methylation in lung tissue may reveal genes and pathways relevant to sex differences in susceptibility, incidence and prevalence of chronic lung diseases.
Materials and methods
Adult lung tissue samples were collected from individuals who underwent thoracic surgery due to lung transplantation, lung volume reduction or lung nodule resection as previously described [Citation32]. All of the participants provided written informed consent. All subjects were former smokers who quit smoking at least one month before surgery. Phenotypic information including demographics, anthropometrics, smoking history and results of pulmonary function testing were obtained from medical records. Severe COPD was defined as GOLD grades 3 or 4, and control smokers had normal spirometric values (forced expiratory volume in 1 second (FEV1) ≥ 80% predicted and ratio FEV1 to forced vital capacity (FVC) ≥ 0.7) [Citation33]. Sex-specific DNA methylation loci in adult lung tissue were compared with those associated in foetal lung tissue. Details about 78 foetal lung tissue samples have been previously described [Citation34]. Foetal lung tissue samples were included to investigate whether sex differences observed in adult lung tissue are detectable during lung development. DNA samples were isolated from discarded foetal lung tissue from 57 to 122 days of gestation, and intrauterine smoking (IUS) exposure was categorized as a binary variable (0: unexposed as reference, 1: exposed subjects) as previously described [Citation34]. We also evaluated replicated associations for lung tissue in a published sex-specific association study of cord blood. All results were modelled on the logit-transformed (M) scale [Citation31]. All the statistical analyses were performed using R version 3.6.0. Subject characteristics are presented as mean (± standard deviation) or median (quartiles) for continuous variables and relative frequencies for categorical variables. Means were compared using t-test and categorical variables were compared using chi-squared test. A P value <0.05 was considered to be statistically significant for demographic comparisons.
DNA methylation assessment
Adult lung tissue samples were frozen and stored at –80°C; DNA was extracted, and 1 µg of extracted DNA from each sample was bisulphite-converted using the EZ DNA Methylation Gold Kit (Zymo Research, Irvine, CA). The genome-wide methylation levels of the DNA samples were assayed using the Infinium HumanMethylation450 BeadChip assay from Illumina (San Diego, CA). A total of 485,512 genome-wide cytosine-phosphate-guanine (CpG) sites were interrogated for each sample, and data importing and pre-processing were performed using R and Bioconductor packages including minfi (version 1.14.0). Detailed methods for quality control have been published [Citation35]; the final number of probes for analysis was 349,826 for 130 white subjects, as previously described [Citation35]. Sex was verified with X chromosome methylation patterns for both adult and foetal lung tissue. The methylation values (β value with a range 0–1) were calculated using β = meth/(meth + unmeth + offset); meth and unmeth are the fluorescence intensity values at methylated versus unmethylated sites, and default Illumina value of offset was 100. Then β values were converted to M values for all analyses to control for heteroscedasticity; results are presented on the ‘beta’ scale as percent methylation to improve interpretability of differential methylation by sex at associated sites. Gene symbols were updated as approved names based on HUGO Gene Nomenclature Committee database (http://www.genenames.org).
Methylation association analysis
Association analysis for each methylation site was performed using R with the Bioconductor package limma (version 3.40.2). Models for sex association with methylation were adjusted for age, presence of COPD, smoking pack-years, time since quitting smoking, centre, and plate numbers. For the interaction analysis between sex and pack-years of smoking or sex and time since quitting smoking, an interaction term with sex and smoking behaviour was added to the previous model for the main effect of sex including age, presence of COPD, centre, plate number, smoking pack-years and time since quitting smoking; there were no suggested technical artefacts related to sentrix position so this was not adjusted for in the model of adult lung tissue. For sex-specific methylation analysis of foetal lung tissue, foetal age, in utero nicotine exposure, sample plate and sentrix position were adjusted for in the model. Nicotine exposure was treated as a dichotomous variable based on placental cotinine [Citation34]. Differentially methylated regions (DMR) were identified using the DMRcate package with parameters including the presence of at least two probes residing within 1000 base-pairs. A False Discovery Rate (FDR) of 5% is applied for the analysis of sex as a predictor of DNA methylation. A less stringent FDR threshold of 20% for the interaction analysis has been applied with presentation of the top 10 sites. Discovery thresholds using FDR < 20% have been previous applied in DNA methylation studies [Citation36–39]. Regional genomic plots containing regulatory information for individual CpG sites of interest were produced using the R package Sushi (version 1.22.0). Regulatory feature data was downloaded (January 2020) from Ensemble BioMart [Citation40] that included information produced from the ENCODE, Roadmap Epigenomics and Blueprint projects [Citation41]. This regulatory feature data described in the Ensembl Regulatory Build documentation were the Enhancer, Promoter Flanking, Promoter, CTCF Binding Site, TF binding site and Open chromatin tracks in the Sushi plot. DNase I Hypersensitivity Clusters in 125 cell types from ENCODE were downloaded from the UCSC database [Citation42]. The Bioconductor package missMethyl (version 3.9) was used to test for enrichment in KEGG pathways which takes into account the number of CpG sites per gene to avoid bias due to the different numbers of CpG sites profiled for each gene [Citation43,Citation44]. Annotation of genes associated with each CpG site was based on data provided by Entrez Gene.
Lung tissue gene expression
RNA was extracted from the same adult lung tissue samples using the AllPrep kit (Qiagen, Valencia, CA), and gene expression profiling was assessed using HumanHT-12 BeadChips (Illumina, San Diego, CA) as previously reported [Citation35,Citation45]. Quality control was performed using quantile, signal-to-noise, correlation matrix and principal component analysis (PCA) to identify outliers and low-quality samples as published previously [Citation45], and Pearson correlation between DNA methylation and gene expression was performed using the cor function without weighting in the stats package in R. Methylation and expression features were linked by gene name and the gene coordinates; all CpGs evaluated were within the gene or within 50 base pairs of the gene coordinates. Information about genomic context are labelled on the scatter plots.
Results
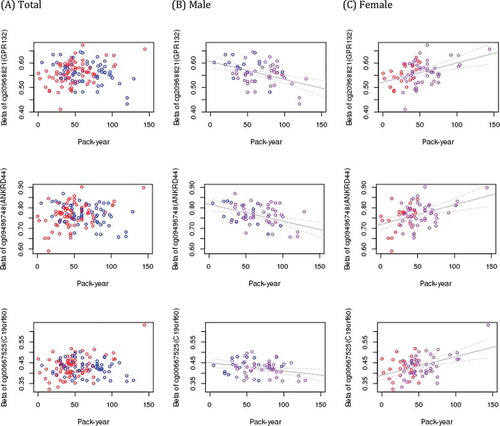
DNA methylation data for adult lung tissue were available from 130 white subjects. Subjects’ median age was 63 (IQR 60, 67.75) years old, 60 (46.2%) were male sex and 92 (70.8%) had COPD. There was no significant difference between males and females by age, prevalence of COPD, FEV1 (%predicted) or FVC (%predicted) and time since quit smoking. Pack-years were significantly higher in males despite similar mean lung function between males and females (). When adjusted for age, presence of COPD, smoking pack-years, time since quitting smoke, centre, and sample plate, sex was associated with 710 differentially methylated sites at FDR < 0.05 (, Table S1, and ). The most significant sex-associated CpG site was cg03691818, mapped to the KRT77 gene and hypo-methylated in males compared to females. Foetal lung tissue demonstrated 1223 sex differentially methylated loci at FDR < 0.05. The most significant sex-associated CpG site was also cg03691818 annotated to KRT77 gene in foetal lung tissue, same as adult lung tissue. Comparing sex-specific methylation loci of adult lung tissue with those of foetal lung and cord blood (FDR < 0.05), found 240 loci overlapping in all 3 tissues, 54 lung tissue-specific loci associated with sex only in adult and foetal lung, and 241 adult and 624 foetal lung tissue-specific loci (Supplemental Figure S1). The effect estimates of the 54 overlapping loci between adult and foetal lung tissues were all in the same direction. The detailed relationship of each locus associated with sex in adult lung and foetal lung tissues are summarized in Table S1. There was no significant site for pack-years or time since quitting smoking associated with adult lung tissue methylation at FDR< 0.05 (Manhattan plots of DNA methylation associations for smoking pack-years and time since quitting smoking are shown in Figure S2). To evaluate potential sex association interactions with smoking we modelled an interaction term between sex and pack-years and between sex and time since quitting smoking. The top 10 CpG sites for sex by pack-year interactions revealed no associations at a strict FDR threshold of 5%; suggestive findings are noted (). The functional annotation plot for each gene is summarized (Figure S3). Among the top 10 sites for sex by pack-years, nominal associations with pack-years were observed at 8 sites in males and 9 sites in females (). These sites were relatively hypo-methylated in men and relatively hyper-methylated in women with increasing of pack-years except cg10338830 (epithelial membrane protein 3: EMP3) and cg03528353 (STE20 Related Adaptor Alpha: STRADA) ( and Figure S4). Among these top 10 sites, cg20968821 (G protein-coupled receptor 132: GPR132), cg09496748 (ankyrin repeat domain 44: ANKRD44), cg10338830 (EMP3), cg10414350 (methionine sulphoxide reductase A: MSRA), cg06776644 (STRADA) and cg08389588 had nominally significant interactions between sex and time since quitting smoking, all in opposite direction of effect compared with pack-years (Table S2). Association with DNA methylation of these sites and time since quitting smoking are plotted in Figure S5. The sites that demonstrate suggested sex interactions in adult lung tissue with smoking did not demonstrate main effects for sex association. For interaction between sex and time since quitting smoking and between sex and COPD (Table S3), there were no significant interactions. From the regional analysis of adult lung tissue, we identified 76 sex-associated differentially methylated regions at Stouffer’s FDR threshold of 5%. The top region consisted of five differentially methylated CpGs and mapped to SNORD43 ()). None of the regions were robust to multiple testing for the sex by smoking interaction model. In foetal lung tissue, the top sex-associated differentially methylated region consisted of 6 differentially methylated CpGs mapped to SNORA69 and ZPBP2 ()). None of the regions was significant for the IUS-exposure interaction model at FDR<0.05. Pathway analysis for 710 sex-specific methylation sites (FDR < 5%) identified 6 nominally significant pathways including the mitophagy pathway (Table S4). Pathway analysis for 294 overall lung tissue sex-specific loci with nominal significance are presented in Table S5. No pathways were enriched at a strict False Discovery Rate of 5%.
Table 1. Baseline characteristics of study population
Table 2. Top 10 CpG sites for differential methylation between sexes
Table 3. Top 10 CpG sites which have interaction between pack-years and sex and their main effects stratified by sex
Table 4. Top 10 DMRs for differential methylation between sexes in (A) adult lung tissue and (B) foetal lung tissue
Figure 1. Manhattan plot (a) and volcano plot (b) of CpG sites differentially methylated by sex (A) The blue line represents the threshold for an FDR of 5%, and the red line represent genome-wide significance cut-off of P = 5 × 10−8. (B) The M value difference of methylation for each CpG site is plotted on the x-axis, and log-transformed P value is plotted on y-axis. Each point represents an individual CpG site. Only top 10 significant sites were annotated with CpG name
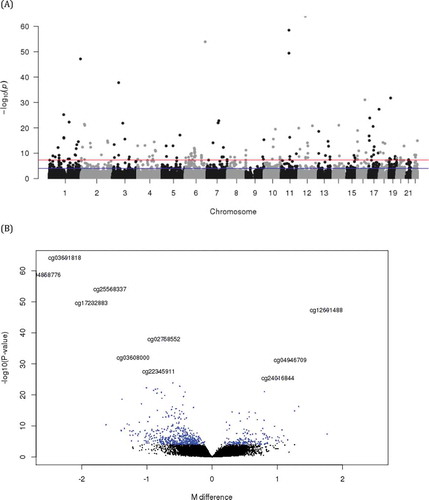
Figure 2. CpG sites with suggested interaction between pack-year of smoking and sex. Blue dots represent male and red dots represent female in the first column (a). In plots of male (b) and female (c), violet dots represent subjects with COPD. The value of x- axis is pack-years of cigarette smoke, and y- axis is fraction of methylation
Association of DNA methylation and gene expression profiling
We evaluated the correlations between DNA methylation and gene expression for annotated genes that were included in top 10 CpGs loci with suggested interactions between sex and pack-years. Among these sites, methylation at cg20968821 annotated to GPR132 (P = 0.047), cg07805999 annotated to C7orf50 (P = 0.018) and cg10338830 annotated to EMP3 (P = 0.008) was correlated with gene expression of the respective genes. For the CpGs in C7orf50 and EMP3 which are respectively located in N-shelf and CpG island, an increase of DNA methylation was associated with a decrease in gene expression. The CpG in the gene GPR132, which is located in open sea, an increase in gene expression was observed (Figure S6).
Discussion
We identified 710 differentially methylated sites associated with sex at FDR<0.05 and suggested interactions between smoking intensity and sex, with most interaction effect estimates demonstrating opposite directions of association between male and female smokers. No significant site associated with pack-years or time since quitting smoking was found at strict false discovery rates, but these opposite associations for DNA methylation and smoking further support sex-specific methylation perturbations as potentially relevant for sex differences in susceptibility to smoking. DNA methylation is a complex phenomenon which connects the influence of genetic and environmental factors and contributes to sex differences in health and disease development [Citation46,Citation47]. DNA methylation is dynamic and is impacted by ageing, disease and environmental exposures including smoking. Sex-specific differences of DNA methylation have been described previously in blood [Citation31] and for gene expression in lung tissue [Citation22,Citation48,Citation49]. More than half of the sites we observed to have sex-specific methylation (415 loci) overlap with sites previously associated in leukocyte DNA [Citation31]. Sex-specific effects of smoking and features of smoking-related lung diseases including COPD have indicated that women may be more susceptible to develop lung damage with the same or less smoke exposure as men [Citation24–30]. Biological differences such as immunological or hormonal determinants have been suggested as possible mechanisms [Citation50,Citation51], with a more recent suggestion that genetic and epigenetic factors may play a role [Citation51–53]. Given these observations, we evaluated interactions between sex and smoking behaviours. Loci with suggested interactions were annotated as GPR132, C19orf60 and ANKRD44; the associated CpG sites overlap with DNase hypersensitivity sites (GPR132 and C19orf60) and enhancer elements (ANKRD44), supporting the importance of future functional work to follow up these sites for sex-specific impact in the lung. DNA methylation of ANKRD44 has been reported to be associated with smoking [Citation21] and asthma phenotypes [Citation54]. Integration of gene expression and DNA methylation revealed GPR132, C7orf50 and EMP3 genes demonstrating correlation. Gene expression of C7orf50 and EMP3 which were respectively located in N-shelf and CpG islands presented negative association with DNA methylation, but GPR132 located in open sea presented positive correlation. The GPR132 gene encodes a member of G protein-coupled receptor superfamily that activates intracellular signal transduction pathways. GPR132 has been reported to be associated with transfusion-related acute lung injury [Citation55]. The EMP3 gene impacts cell proliferation and cell-to-cell interaction, but the mechanisms are still poorly understood [Citation56]. Sex-specific associations of these genes in the lung have not been described previously.
Pathway analysis of the methylation marks associated with sex did not reveal enrichment of pathways at a strict False Discovery Rate. However, nominal association in the mitophagy pathway was observed and this pathway is a biologically plausible driver of sex differences in lung diseases, including COPD. In a previous study of sex differences in gene regulatory networks, we revealed significant differential targeting of mitochondrial functional pathways as a potential driver of sex differences in COPD [Citation57]. More recently, urine mitochondrial DNA has been suggested as a sex-specific biomarker of COPD associated with worse spirometry and emphysema in men and worse respiratory symptoms in women [Citation58]. Sex-specific regulation of mitochondrial pathways have been observed in cardiovascular disease [Citation59] and our findings may support a similar relevance of mitochondrial pathways in sex-specific features of lung health and disease; more research is needed to support this consideration.
One limitation of this analysis is that there is potential for confounding related to adult lung disease. We adjusted our model for the presence or absence of chronic obstructive lung disease measured by lung function and replicated our findings in foetal lung tissue. We do not have information about other comorbidities. Larger studies of lung tissue from well phenotyped cohorts are needed to confirm our findings. Additionally, our study only included former smokers limiting comparison with current or never smokers, and the severity of included COPD subjects was high. There could be recall bias for history of self-reported smoking metrics or exposures other than smoking. Overlapping methylation marks may suggest cellular heterogeneity by sex or blood contamination in the tissue homogenates or true sex divergent signatures that are not tissue specific. We did not observe significant sex-specific gene expression of our loci, although there were several genes having correlations between DNA methylation and gene expression. Differential gene expression by sex has been suggested to be limited, but differential targeting of genes via an epigenetic pathway is likely more relevant [Citation22,Citation58]. Additionally, ascertainment of this tissue cohort may limit generalizability to the general population and to other races. Although we do not have a lung tissue replication set with extensive phenotypic and exposure information, we do demonstrate potential functional relevance of our observations using contemporaneous gene expression data and evaluation of sex-specific association in foetal lung tissue. Lastly, cellular heterogeneity may be relevant to our detection of sex-specific associations; further studies using a single lung cell type will be an important direction for future modelling of sex-specific features of lung disease. Overall, the lung-specific sex divergent methylation marks further support a role for gene regulatory pathways in the lung [Citation58] as important pathways for further study in sex differences in lung health and disease; the epigenome may be particularly informative to study in this regard.
Our findings demonstrated sex differences in DNA methylation of lung tissue which may have relevance to the sex divergence observable in smoking-related lung diseases. We also identified a subset of sex-specific methylation marks with suggested smoking interactions. Sex differences in susceptibility to lung damaging effect of cigarette smoking may be related to epigenetic effects in mitochondrial pathways. Future research on sex-specific aspects of the epigenome may reveal new insights into COPD pathogenesis.
Authors’ contribution
HKK, DLD, JM and PK conceived of the analysis. HKK performed all analysis. DLD and EKS supervised the research project. HKK and DLD drafted the manuscript. JM, KT, STW, CPH and EKS provided substantive review and edited to the manuscripts. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Institutional Review Board approval was obtained at the three centers (Brigham and Women’s Hospital (Boston, MA), St. Elizabeth’s Hospital (Boston, MA) and Temple University Hospital (Philadelphia, PA)) for adult lung tissue dataset. Human fetal lung tissues were obtained from two NICHD-supported tissue retrieval programs at the University of Washington Center for Birth Defects Research (Seattle, WA) and the University of Maryland Brain and Tissue Bank for Developmental Disorders (Baltimore, MD). The Institutional Board Review at Brigham and Women’s Hospital declared the use of these tissues non-human subject research (2010-P-002399).
Supplemental Material
Download MS Word (1.4 MB)Supplemental Material
Download MS Excel (84.2 KB)Disclosure statement
DLD reports grants support from Novartis and Bayer. EKS has received grant support from GlaxoSmithKline and Bayer. CPH reports grant support from Boehringer-Ingelheim, Novartis and Bayer. STW reports honoraria from UpToDate
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Vernooy JH, Kucukaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166:1218–1224.
- Willemse BW, Ten Hacken NH, Rutgers B, et al. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26:835–845.
- Conen D, Everett BM, Kurth T, et al. Smoking, smoking cessation, [corrected] and risk for symptomatic peripheral artery disease in women: a cohort study. Ann Intern Med. 2011;154:719–726.
- Kawachi I, Colditz GA, Stampfer MJ, et al. Smoking cessation and decreased risk of stroke in women. JAMA. 1993;269:232–236.
- Newcomb PA, Carbone PP. The health consequences of smoking—cancer. Med Clin North Am. 1992;76:305–331.
- Vineis P, Alavanja M, Buffler P, et al. Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst. 2004;96:99–106.
- Ezzati M, Lopez AD. Estimates of global mortality attributable to smoking in 2000. Lancet. 2003;362:847–852.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442.
- Rodgman A, Perfetti TA. The chemical components of tobacco and tobacco smoke. 2nd ed. Boca Raton: CRC Press; 2013.
- DeMeo DL, Ramagopalan S, Kavati A, et al. COPDGene investigators. Women manifest more severe COPD symptoms across the life course. Int J Chron Obstruct Pulmon Dis. 2018 Oct;1(13):3021–3029.
- Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132.
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
- Zeilinger S, Kühnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013 May 17;8(5):e63812.
- Tsaprouni LG, Yang TP, Bell J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014 Oct;9(10):1382–1396.
- Sayols-Baixeras S, Lluís-Ganella C, Subirana I, et al. Identification of a new locus and validation of previously reported loci showing differential methylation associated with smoking. The REGICOR study. Epigenetics. 2015;10(12):1156–1165.
- Guida F, Sandanger TM, Castagné R, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015 Apr 15;24(8):2349–2359.
- Qiu W, Wan E, Morrow J, et al. The impact of genetic variation and cigarette smoke on DNA methylation in current and former smokers from the COPD Gene study. Epigenetics. 2015;10(11):1064–1073.
- Ambatipudi S, Cuenin C, Hernandez-Vargas H, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016 May;8(5):599–618.
- Zhu X, Li J, Deng S, et al. Genome-wide analysis of DNA methylation and cigarette smoking in a Chinese population. Environ Health Perspect. 2016 Jul;124(7):966–973.
- Wilson R, Wahl S, Pfeiffer L, et al. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics. 2017 Oct 18;18(1):805.
- McCartney DL, Stevenson AJ, Hillary RF, et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine. 2018 Nov;37:214–220.
- Yang CX, Shi H, Ding I, et al. Widespread sexual dimorphism in the transcriptome of human airway epithelium in response to smoking. Sci Rep. 2019 Nov 26;9(1):17600.
- Zhou J, Sears RL, Xing X, et al. Tissue-specific DNA methylation is conserved across human, mouse, and rat, and driven by primary sequence conservation. BMC Genomics. 2017 Sep 12;18(1):724.
- Chen Y, Horne SL, Dosman JA. Increased susceptibility to lung dysfunction in female smokers. Am Rev Respir Dis. 1991;143:1224e30.
- Gan WQ, Man SF, Postma DS, et al. Female smokers beyond the perimenopausal period are at increased risk of chronic obstructive pulmonary disease: a systematic review and meta-analysis. Respir Res. 2006;7:52.
- Langhammer A, Johnsen R, Gulsvik A, et al. Sex differences in lung vulnerability to tobacco smoking. Eur Respir J. 2003;21:1017–1023.
- Prescott E, Bjerg AM, Andersen PK, et al. Gender difference in smoking effects on lung function and risk of hospitalization for COPD: results from a Danish longitudinal population study. Eur Respir J. 1997;10:822–827.
- Xu X, Li B, Wang L. Gender difference in smoking effects on adult pulmonary function. Eur Respir J. 1994;7:477–483.
- Xu X, Weiss ST, Rijcken B, et al. Smoking, changes in smoking habits, and rate of decline in FEV1: new insight into gender differences. Eur Respir J. 1994;7:1056–1061.
- Silverman EK, Weiss ST, Drazen JM, et al. Gender-related differences in severe, early-onset chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162:2152–2158.
- Yousefi P, Huen K, Davé V, et al. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genomics. 2015;16:911.
- Morrow JD, Cho MH, Hersh CP, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics. 2016;11(10):730–739.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global Strategy For The Diagnosis, Management And Prevention of COPD; cited 2019 Aug. Available from: http://goldcopd.org/download/326/
- Chhabra D, Sharma S, Kho AT, et al. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics. 2014 Nov;9(11):1473–1484.
- Morrow JD, Cho MH, Platig J, et al. Ensemble genomic analysis in human lung tissue identifies novel genes for chronic obstructive pulmonary disease. Hum Genomics. 2018 Jan 15;12(1):1.
- Benton MC, Johnstone A, Eccles D, et al. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015;16(1):8.
- Hannon E, Dempster E, Viana J, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176.
- Wong Doo N, Makalic E, Joo JE, et al. Global measures of peripheral blood-derived DNA methylation as a risk factor in the development of mature B-cell neoplasms. Epigenomics. 2016;8:55–66.
- Morrow JD, Make B, Regan E, et al. DNA methylation is predictive of mortality in current and former smokers. Am J Respir Crit Care Med. 2020 May 1;201(9):1099–1109.
- Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440.
- Zerbino DR, Wilder SP, Johnson N, et al. The ensembl regulatory build. Genome Biol. 2015;16:56.
- Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013;41:D56–D63.
- Geeleher P, Hartnett L, Egan LJ, et al. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics. 2013 Aug 1;29(15):1851–1857.
- Wang Z, Wu X, Wang Y. A framework for analyzing DNA methylation data from Illumina Infinium HumanMethylation450 BeadChip. BMC Bioinformatics. 2018 Apr 11;19(Suppl 5):115.
- Morrow JD, Zhou X, Lao T, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep. 2017 Mar;13(7):44232.
- Cunningham CM, Eghbali M. An introduction to epigenetics in cardiovascular development, disease, and sexualization. Adv Exp Med Biol. 2018;1065:31–47.
- Xiao FH, Chen XQ, He YH, et al. Accelerated DNA methylation changes in middle-aged men define sexual dimorphism in human lifespans. Clin Epigenetics. 2018;10(1):133.
- Dugo M, Cotroneo CE, Lavoie-Charland E, et al. Human Lung tissue transcriptome: influence of sex and age. PLoS One. 2016;11(11):e0167460.
- van den Berge M, Brandsma CA, Faiz A, et al. Differential lung tissue gene expression in males and females: implications for the susceptibility to develop COPD. Eur Respir J. 2019 Jul 18;54(1):1702567.
- Jenkins CR, Chapman KR, Donohue JF, et al. Improving the Management of COPD in Women. Chest. 2017 Mar;151(3):686–696.
- Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016 Oct;16(10):626–638.
- Patsopoulos NA, Tatsioni A, Ioannidis JP. Claims of sex differences: an empirical assessment in genetic associations. JAMA. 2007 Aug 22;298(8):880–893.
- Jenny van Dongen J, Nivard MG, Willemsen G, et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun. 2016 Apr 7;7:11115.
- Nicodemus-Johnson J, Myers RA, Sakabe NJ, et al. DNA methylation in lung cells is associated with asthma endotypes and genetic risk. JCI Insight. 2016 Dec 8;1(20):e9015155.
- Ellison MA, Ambruso DR, Silliman CC. Therapeutic options for transfusion related acute lung injury; the potential of the G2A receptor. Curr Pharm Des. 2012;18(22):3255–3259.
- Jun F, Hong J, Liu Q, et al. Epithelial membrane protein 3 regulates TGF-β signaling activation in CD44-high glioblastoma. Oncotarget. 2017;8(9):14343–14358.
- Glass K, Quackenbush J, Silverman EK, et al. DeMeo DL Sexually-dimorphic targeting of functionally-related genes in. COPD BMC Syst Biol. 2014 Nov 28;8:11.
- Zhang WZ, Rice MC, Hoffman KL, et al. SPIROMICS investigators. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight. 2020 Feb 13;5(3):e133984.
- Vona R, Ascione B, Malorni W, et al. Mitochondria and sex-specific cardiac function. Adv Exp Med Biol. 2018;1065:241–256.