
ABSTRACT
To assess the m6A methylome in the anterior capsule of the lens of high myopia patients. MeRIP-seq and RNA-seq were performed to identify differences in the m6A methylomes and gene expression between anterior capsule of the lens of simple nuclear cataract patients (N) and nuclear cataract patients with high myopia (G). Expression of m6A-related enzymes was confirmed by quantitative real-time-PCR. ALKBH5 was downregulated in G. The observed m6A peak was identical to the conserved RRACH gmotif and was markedly correlated with two distinct coordinates. Differentially methylated genes were enriched in some pathways regulating the formation of extracellular matrix. These findings suggest that upregulation of m6A methylation may change fundus anatomy by regulating the composition of the extracellular matrix through encoding protein.
Introduction
With the change in lifestyle, great modifications have taken place in human eye-use habits, and the incidence of high myopia is increasing. At present, 163 million of people, who account for 2.7% of the world total population, are suffering from high myopia [Citation1–3]. Worryingly, this number is rising sharply [Citation2–4]. High myopia, defined as myopia exceeding 6.00 dioptres or axis length ≥26 mm, is a disorder that affects almost the entire human eye, from the anterior pole to the posterior pole, including high ametropia, cataract, open-angle glaucoma, and retinopathy [Citation5]. Excessive axial elongation of the eye in high myopia can cause mechanical stretching of the outer coats of the eyeball, resulting in various pathologic changes such as staphyloma, chorioretinal atrophic lesions, lacquer cracks, and choroidal neovascularization.
N6-methyladenosine (m6A), as the most prevalent internal form of modification in polyadenylated mRNAs and long noncoding RNAs in higher eukaryotes, was first identified in the 1970s [Citation6]. Studies in bioscience and medicine have revealed that RNA m6A plays important biological roles in the regulation of cellular metabolic processes. In 2014, Batista et al. have found that RNA m6A controls cell transition fate in mammalian embryonic stem cells [Citation7]. The next year, Zhao et al. have discovered that RNA m6A regulates pluripotency in murine stem cells [Citation8]. In addition, Shen et al. have studied the role of m6A in shoot stem cell fate in Arabidopsis in 2016 [Citation9]. Furthermore, m6A methylation plays important roles in human disease such as control of HIV-1 replication and interaction with the host immune system during HIV-1 infection of T cells [Citation10], promoting translation of oncogenes in human lung cancer [Citation11] and induction of breast cancer stem cell phenotype [Citation12]. Thus, more medical fields will reveal the role of m6A methylation. In mammals, m6A methylation modification involves three enzymes: methylase (METTL3, METTL14, and WTAP), demethylase (FTO and ALKBH5), and methylation recognition enzyme (YTHDF1, YTHDF2, and YTHDF3). Enzyme abnormality causes a series of diseases, including tumour, neurological diseases, and circulatory disorders [Citation13–15]. However, the role of m6A methylation in high myopia is still unclear.
To investigate the functions of m6A, facilitate future studies of mammalian m6A, and explore the pathogenic mechanism of the eye caused by high myopia, we detected the m6A methylomes of anterior lens capsule in high myopia patients. This enabled us to acquire a set of transcriptome-wide m6A profiles in patients with high myopia and investigate tissue and breed generality and selectivity of methylated genes and their functional implications.
Materials and methods
Acquisition of biological samples
Twelve anterior lens capsule samples were enrolled and divided into two groups. One group was characterized by simple nuclear cataract patients (N, 6 samples), while the other group was characterized by nuclear cataract patients with high myopia (G, 6 samples). Each patient underwent conventional cataract phacoemulsification combined with intraocular lens implantation. During continuous circular capsulorhexis, the anterior capsule was collected into a cryopreservation tube and stored in −80ºC. Patients with nuclear cataract and an axis length ≥29 mm were included in this study. Whereas patients with other eye diseases and other systemic diseases such as hypertension, diabetes, coronary heart disease, and autoimmune disease were excluded from the investigation. Patient information is shown in . Informed consent was obtained from all individual participants included in the study. All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional and National Research Committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Table 1. Patient characteristics of G and N
Preparation and sequencing of RNA library
RNA high-throughput sequencing was performed by Cloud-Seq Biotech (Shanghai, China). Briefly, total RNA was used for removing the rRNAs using the NEBNext® rRNA Depletion kit (New England Biolabs, Inc., MA, USA) following the manufacturer’s instructions. RNA libraries were constructed by using NEBNext® Ultra™ II Directional RNA Library Prep kit (New England Biolabs) according to the manufacturer’s instructions. Libraries were controlled for quality and quantified using the Bioanalyzer 2100 system (Agilent Technologies, Inc., USA). Library sequencing was performed on an illumina Hiseq instrument with 150 bp paired-end reads.
Preparation and sequencing of MeRIP library
m6A RNA-seq service was provided by Cloud-seq Biotech Inc. Briefly, m6A RNA immunoprecipitation was performed using the GenSeq™ m6A RNA IP kit (GenSeq Inc., China) by following the manufacturer’s instructions. The quality and quantity of total RNA were assessed by using NanoDrop ND-2000 (Thermo Fisher Scientific, USA). RNA purity was qualified when OD260/OD280 values ranged from 1.8 to 2.1. RNA integrity and gDNA contamination were measured using denatured agarose gel electrophoresis. Agilent 2100 Bioanalyzer was used to detect library quality. RNA quantification and quality assurance were assessed using Qubit3. Both the input sample without immunoprecipitation and the m6A IP samples were used for RNA-seq library generation using NEBNext® Ultra II Directional RNA Library Prep kit (New England Biolabs). The library quality was evaluated using the BioAnalyzer 2100 system (Agilent Technologies, Inc.). Library sequencing was performed on an illumina Hiseq instrument with 150 bp paired-end reads.
Data analysis
Data analysis of mRNA sequencing results
Paired-end reads were obtained from the Illumina HiSeq 4000 sequencer and were quality-controlled using Q30. After 3ʹ adaptor-trimming and low-quality reads removing using the cutadapt software (v1.9.3) [Citation16], the high-quality clean reads were aligned to the reference genome (UCSC hg19) using the Hisat2 software (v2.0.4) [Citation17]. Guided by the Ensembl gtf gene annotation file, the cuffdiff software [Citation18] (part of cufflinks) was used to get the gene-level FPKM as the expression profiles of mRNA, fold change and p-value were calculated based on FPKM, and differentially expressed mRNA were identified. Gene ontology (GO) and pathway enrichment analysis were performed based on the differentially expressed mRNAs. GO and pathway enrichment analysis were performed using the Database for Annotation, Visualization, and Integrated Discovery [Citation19,Citation20]. The ontology covers three parts: cellular component (CC), molecular function (MF), and biological process (BP). The p-value denotes the significance of GO term enrichment of the genes. Pathway enrichment analysis is a functional analysis that maps genes to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The Fisher p-value denotes the significance of the pathway correlated to the conditions.
Data analysis of MeRIP sequencing results
Briefly, paired-end reads were obtained from the Illumina HiSeq 4000 sequencer and were quality-controlled using Q30. Then, 3ʹ adaptor-trimming and low quality reads removing using the cutadapt software (v1.9.3) [Citation16] were performed. Clean reads of all libraries were aligned to the reference genome (HG19) using the Hisat2 software (v2.0.4) [Citation17]. Reads were matched to the results on the genome (bam file), and the IGV software [Citation21] was used for visualization to observe the abundance of reads at specific locations in the genome of each sample. Methylated sites on RNAs (peaks) were identified using the MACS software [Citation22]. Differentially methylated sites were identified using diffReps [Citation23]. These peaks identified by both software overlapping with exons of mRNA were identified and selected using home-made scripts. GO and pathway enrichment analysis were performed for the differentially methylated protein-coding genes. MEME software was used for motif analysis [Citation24].
RNA extraction and quantitative real-time PCR
Levels of the mRNA m6A-related genes METTL3, METTL14, FTO, ALKBH5, YTHDF1, and YTHDF2 were analysed in G and N groups. TRIzol reagent (Invitrogen, USA) was used to isolate total RNA, which was then used to synthesize complementary DNA by using the SuperScript™ III Reverse Transcriptase (Invitrogen). Real-time PCR was performed by using qPCR SYBR Green Master Mix (CloudSeq, Shanghai) and QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific, USA). All procedures were performed according to the manufacturer’s protocols. The sequences of primers used are presented in .
Table 2. Primers used for quantitative real-time-PCR
Statistical analyses
The significance of differences between G and N groups was tested by using unpaired two-tailed Student’s t-test. All statistical analyses were conducted using GraphPad Prism v7.00 software. The differences were considered significant if p-value was <0.05.
Results
Overview of m6A methylation map in anterior capsular of G lens
We calculated the methylation sites in the two groups and found that the number of methylation sites in the G group was higher than that in the N group ().
Figure 1. (a) The number of methylation sites and genes of G and N. (b)The methylation difference between N and G. (c) Distribution of differentially methylated m6A sites with significance in chromosomes of high myopia
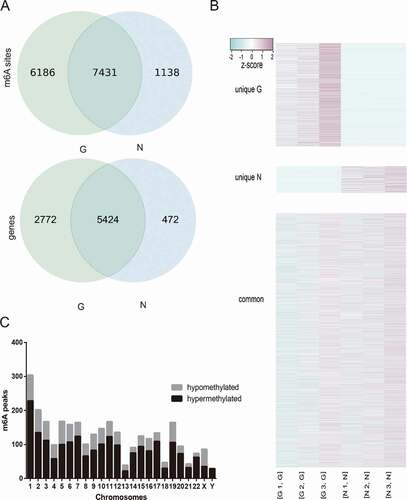
More than 80% and 70% of the m6A peaks were consistently detected in three biological replicates of N and G groups, respectively. These recurrent peaks should be regarded as highly enriched m6A peaks for further analysis. The results showed that 13,617 m6A recurrent peaks among 8,196 expressed genes in N and 8,569 m6A recurrent peaks among 5896 expressed genes in G were detected (). We performed genome-wide profiling of m6A-modified mRNA in G and N. Compared to N, 2,083 significantly hypermethylated m6A peaks and 907 significantly hypomethylated m6A peaks were identified in G (fold change ≥2 and p ≤ 0.0001). The top 20 altered m6A peaks are listed in . The hypermethylated m6A peaks were identified in all chromosomes (especially chr1), while no hypomethylated m6A peak was found in chrY ().
Table 3. The top 20 differently methylated m6A peaks
To understand the preferential location of m6A in mRNA, we investigated the metagene profiles of m6A peaks in the mRNA transcriptome. m6A peaks were markedly correlated with two distinct coordinates: immediately following near the end of the 5ʹ untranslated regions (5ʹUTRs) and start of the coding sequence (CDS) and near the end of the CDS and the beginning of the 3ʹ untranslated region (3ʹUTRs) ().
Figure 2. (a) Preferential location of m6A in mRNA. Each transcript is divided into three parts including 5ʹuntranslated region, coding DNA sequence and 3ʹuntranslated region. (b) Pie charts showing m6A peaks distribution in different gene context. (c) Data visualization of MYL12A mRNA m6A modification in G
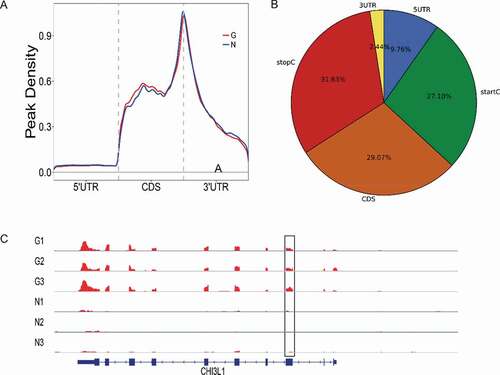
The stop codon of peaks was more pronounced than the start codon of peaks (31.63% vs 27.10%). To assess the enrichment methodically, we assigned each m6A peak to one of the five non-overlapping transcript segments: 5ʹUTRs, start codon, CDS, stop codon, and 3ʹUTR. As a result, 87% of m6A peaks were within genic regions, and more than 60% of genic peaks were localized near the stop codon and CDS, while 12% were found in the 5ʹUTRs and 3ʹUTRs (). The topological patterns distributing within genes were highly similar in both samples, suggesting that recognition of motif for m6A methylation was conserved among anterior capsule of human lens. MYL12A, a considerably hypermethylated peak, is shown in .
Notably, more than 3,500 of m6A-methylated coding genes contained only one m6A peak, while a relatively small number of genes contain two or more peaks (), which was consistent with the trend of the proportions previously reported in the mouse brain and pig liver [Citation25].
Figure 3. (a) Number of m6A peaks per gene. more than 3500 m6A-methylated coding genes contained only one m6A peak. (b) The top five motifs enriched across m6A peaks identified from G
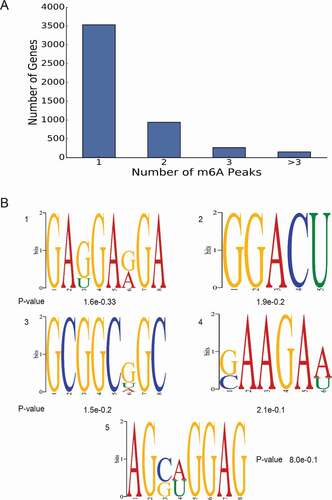
An unbiased search for motifs enriched in regions surrounding m6A peaks was performed to determine whether the identified m6A peak was identical to the conserved RRACH motif (where R represented purine, A was m6A, and H was a non-guanine base) [Citation26,Citation27]. Clustering of significantly enriched sequences perfectly confirmed the previously established m6A RRACH consensus sequence in both groups (). The identification of a strong consensus reinforces the authenticity of the discovered m6A peaks and supports the existence of a predominant methylation machinery.
m6A-containing genes are involved in important biological pathways
To further determine general biological functional pathways that the significance of m6A methylation in high myopia development, the genes containing significantly altered m6A peaks (differentially methylated genes, G) were analysed by performing GO and KEGG pathway analysis. We systematically screened these different and common peaks and the related genes and identified the GO terms with the help of the GO consortium database ().
Figure 4. (a–c) Major gene ontology terms were significantly enriched for the hypermethylated genes. (d–f) Major gene ontology terms were significantly enriched for the hypomethylated genes
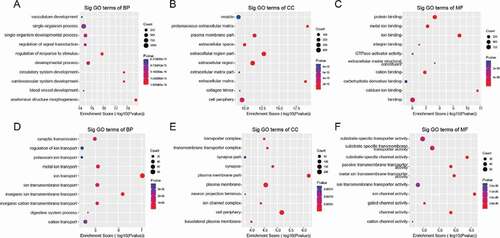
GO analysis showed that the hypermethylated m6A peak-related genes encoding m6A-containing mRNAs were mainly involved in a variety of biological processes, including anatomical structure morphogenesis (ontology: biological process; ), proteinaceous extracellular matrix (ontology: cellular component; ), extracellular matrix structural constituent (ontology: molecular function; ). The hypomethylated genes were significantly associated with regulation of ion transport (ontology: biological process; ), plasma membrane part (ontology: cellular component; ), and ion channel activity (ontology: molecular function; ). GO biological process classification indicated that most of the m6A methylation was enriched in the cellular process (). These genes were involved in the formation of extracellular matrix (), suggesting that extracellular matrix was associated with the formation of pathological damage to high myopia.
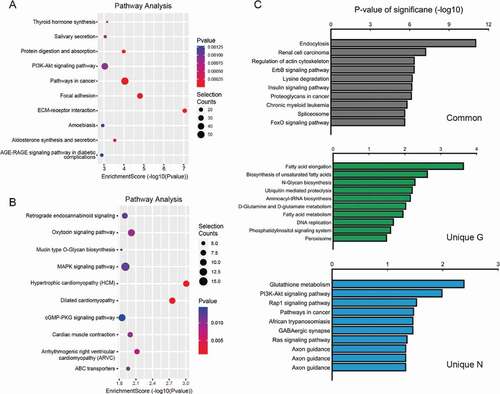
Outstandingly, KEGG pathway analysis showed the result of unique G and unique N (), in addition to information on hypermethylated genes () and hypomethylated genes ().
Figure 5. (a) The top ten significantly enriched pathways for the hypermethylated genes. (b) The top ten significantly enriched pathways for the hypomethylated genes. (c) Unique pathway of G and N
Conjoint analysis of RNA-seq and MeRIP-seq
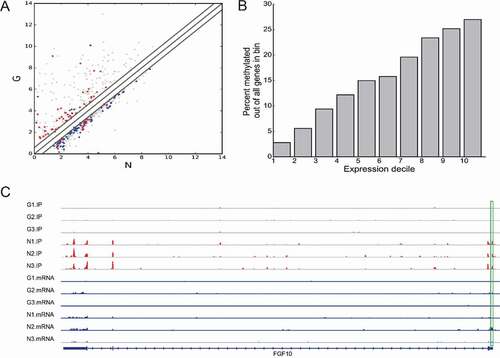
Transcriptome profiles of altered genes in the anterior capsule of patients with high myopia were determined using RNA-seq and shown in . Compared to N, 20,385 genes were differentially expressed in G (fold change ≥1.5 and p < 0.05), including 7,834 upregulated genes and 1,2524 downregulated genes. The top 20 altered genes are listed in . The top 10 GO and KEGG pathways are shown in Supplementary Figures 1 and 2. Based on conjoint analysis of differentially expressed genes and differentially methylated genes, the higher the transcription expression, the higher the methylation ratio ().
Figure 6. (a) Scatter plots showing the differentially expressed genes (fold changes ≥2 and p < 0.05). (b) m6A and gene expression of transcript. (c) Data visualization of methylation and gene expression
ALKBH5 and FTO were downregulated in G
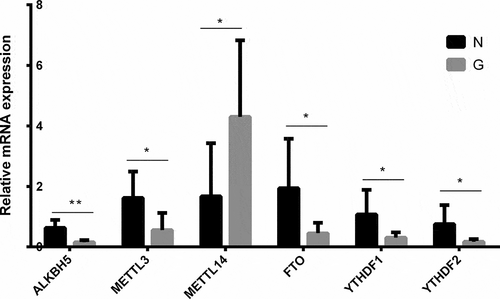
Based on quantitative real-time-PCR, the expressions of six major enzymes involved in m6A methylation, including METTL3, METTL14, FTO, ALKBH5, YTHDF1, and YTHDF2, were verified between G and N. As shown in , in G, the levels of demethylases ALKBH5 and FTO, demethylases acting to remove m6A modification, significantly decreased compared to those in N (ALKBH5:G/N = 0.245, P < 0.05; FTO:G/N = 0.234, P < 0.05), whereas METTL3 and METTL14, the key methyltransferases responsible for m6A modifications, significantly decreased (G/N = 0.346, P < 0.05) and increased (G/N = 2.565, P < 0.05), respectively. The levels of two methylation recognition enzymes (YTHDF1 and YTHDF2) significantly decreased in G compared to N (YTHDF1: G/N = 0.284, P < 0.05; YTHDF2: G/N = 0.226, P < 0.05).
Figure 7. The expressions of six major enzymes involved with m6A methylation including METTL3, METTL14, FTO, ALKBH5, YTHDF1, and YTHDF2 between G and N
Expression of candidate genes correlated with pathological damage in high myopia
To further investigate the mechanisms of pathological damage in high myopia, we performed statistical analysis of differentially expressed m6A methylation-modified genes, including C11orf96 (uncharacterized protein), CSF1 (macrophage colony-stimulating factor 1), PTP4A3 (protein tyrosine phosphatase type IVA 3), COL6A3 (collagen alpha-3(VI) chain), CHI3L1 (chitinase-3-like protein 1), PXDN (peroxidasin homolog). However, COL6A3, CHI3L1, and PXDN were most likely to be our target genes.
Discussion
High myopia is a blinding disease with a high incidence, which seriously affects the quality of life of patients. In this study, we obtained the first high myopia transcriptome-wide m6A modification profile using MeRIP-Seq and discovered that mRNA m6A sites were mainly enriched around stop codons, CDS, and 3ʹUTRs, consistent with the distribution characteristics of mammal transcriptomes, although the modifications also occurred in 5ʹUTRs [Citation7,Citation25,Citation28]. The m6A distribution in our study was the typical m6A topological pattern in most of the mature mRNA. The extensively higher m6A signals at the stop codon or 3ʹUTRs contributed to RNA stability, signalling for transport and translocation, or acted as regulatory elements for protein translation through the recruitment of specific factors onto the m6A sites for RNA transport or protein synthesis [Citation29,Citation30]. High-throughput m6A RNA sequencing databases showed that the distribution of m6A modifications on mRNA was sequence-specific and tended to occur in the conserved motif RRACH [Citation25,Citation28,Citation31–35]. Accordingly, in our study, we successfully identified the consensus motif sequence in patients with high myopia transcriptome.
m6A mRNA modifications can be dynamically regulated by a multicomponent methyltransferase complex with opposite modifying activities, including m6A writers, m6A erasers, and m6A readers. Increasing evidence suggests that m6A modification plays a significant role in the proliferation, migration, and invasion of tumour cells. METTL3 was reported to be an oncogene in human lung cancer cells by promoting the translation of certain mRNAs such as epidermal growth factor receptor and Hippo pathway effector TAZ [Citation11]. A study verified that METTL14 interacted with the microprocessor protein DGCR8 and positively modulated the primary microRNA 126 process in an m6A-dependent manner [Citation36]. In ophthalmology, Jia et al. have found that YTHDF1 suppresses ocular melanoma by modulating HINT2 mRNA translation [Citation37]. Luo et al. have confirmed that silencing METTL3 significantly suppressed uveal melanoma cell proliferation and colony formation through cell cycle G1 arrest, as well as migration and invasion [Citation38]. Recently, it was reported that METTL3 silencing promoted the proliferation and alleviated the apoptosis of high glucose-induced human lens epithelial cells [Citation39]. These studies have proved that RNA m6A methylation also plays an important role in the development of ophthalmic diseases. In our study, METTL14 was upregulated and METTL3, FTO, and ALKBH5 were downregulated in the anterior capsule of high myopia patient lens as determined using qPCR. This inconsistency of changes disrupts the dynamic equilibrium of writers and erasers of m6A modification. Our data suggest that m6A methylation is strongly associated with the pathogenic mechanism of high myopia.
RNA m6A methylation plays a key role in the regulation of post-transcriptional gene expression [Citation25,Citation31,Citation40,Citation41]. The importance of m6A in post-transcriptional regulation of gene expression is further reinforced by the discovery and characterization of mammalian reader proteins that recognize m6A modifications of mRNA and subsequently affect the stability of the target transcripts. YTHDF1 and YTHDF2, members of YTH domain-containing proteins, closely resembled each other and were predominantly cytoplasmic, but their mechanisms could be different. YTHDF2 mediated the decay of target mRNAs, whereas YTHDF1 interaction with eIF3 and other translation initiation factors suggested that it might affect translation rather than affect the half-lives of mRNAs [Citation42,Citation43]. In our study, compared to N, we confirmed that the YTHDF1 and YTHDF2 were downregulated in G. This result indicated that two readers might participate in the progress of high myopia through regulation of post-transcriptional gene expression. The gene FGF10 (), as a differentially methylated region, may have a certain impact on its downstream proteins, including nElavl and RBM10, which are RNA-binding proteins that directly interact with target RNAs and regulate several aspects of RNA metabolism. Studies have reported that nELAV proteins are unique to neurons and associated with Alzheimer’s disease [Citation44]. Therefore, we speculate that hypomethylated FGF10 may enhance the ability to bind nELAV protein, playing an important role in the mechanism of high myopia brain injury. RBM10, another FGF10-binding protein, promotes many transformation and hypoxia-associated processes and events, including angiogenesis [Citation45–47]. The occurrence of choroidal neovascularization in high myopia may be related to hypomethylation, weakening the ability of FGF10 to bind to RBM10.
One of the main functions of m6A is to mediate mRNA degradation in mammalian cells [Citation29,Citation48–50], suggesting a possible negative relationship between the m6A methylation extent and the transcript level. However, this observation somewhat differed from our present result in the anterior capsule of human lens, which showed that most of the highly expressed transcripts were relatively more modified by m6A. These differences may be due to different methodologies, different biological species, or different tissue samples. The results further indicated that different tissues might possess different characteristics in m6A methylation sites, suggesting a regulatory role for m6A in gene expression.
Differential m6A methylation has proved responsible for tissue or organ differentiation and development. Genes encoding m6A-containing RNAs in adult mouse brain tissue are linked to neurodevelopmental and neurological disorders [Citation25,Citation31]. Moreover, m6A RNA methylation in Drosophila and Zebrafish early embryogenesis shows a conserved mechanism of neuronal mRNA regulation contributing to brain function [Citation43]. Our study uncovers regulation roles of m6A modification in the anterior lens capsule of high myopia patients. Differentially methylated genes mostly participate in the regulation of anatomical structure morphogenesis pathways. GO analysis showed that the upregulated m6A peak-related genes encoding m6A-containing mRNAs were mainly involved in the formation of extracellular matrix, consistent with the results shown by Wang [Citation51]. In differentially expressed m6A methylation-modified genes, CHI3L1 (chitinase-3-like protein 1), encoding protein YKL-40, is thought to play a role in tissue remodelling and in the capacity of cells to respond to and cope with changes in their environment [Citation52]. In addition, YKL-40 is an inflammatory marker that plays an important role in pathological processes such as cell proliferation, migration, differentiation, and tissue remodelling of inflammatory reactions [Citation53–55]. Therefore, we suspect that the upregulation of methylase (METTL14) and the downregulation of demethylase (FTO and ALKBH5) catalyse the hypermethylation of gene CHI3L1, which may affect the expression level of its encoded protein YKL-40 and promotes the pathological state of high myopia by regulating the composition of the extracellular matrix. Posterior scleral staphyloma is one of the most basic changes in fundus anatomy in a series of interrelated degenerative changes in high myopia [Citation56]. Thus, high expression of m6A methylation of mRNA may be involved in the formation of posterior scleral staphyloma by affecting the components of the extracellular matrix.
The choroid circulation receives approximately 95% of blood from the ophthalmic artery and provides oxygen and nutrition to the outer retinal layers. Therefore, the choroidal circulation may play an important role in the retinal dysfunction and vision loss. GO analysis showed that the upregulated m6A peak-related genes encoding m6A-containing mRNAs were also associated with circulatory system development, blood vessel development, and vasculature development (). This suggests that the increase in methylation level of mRNA may cause damage to the fundus of patients with high myopia by affecting the choroidal circulation.
Conclusion
For the first time, we provide the integral human lens transcriptome m6A map in high myopia patients. Our map reveals features of m6A distribution in the high myopia of human transcriptome and identifies generality as well as selectivity of methylated genes and their functional implications. Additionally, the abnormal expression of methylation-related enzymes destroys the dynamic homoeostasis of methylation. This comprehensive methylome profiling provides a solid basis for the determination of potential functional roles for RNA m6A modification in pathological damage to the eye caused by high myopia.
Supplemental Material
Download TIFF Image (434.7 KB)Supplemental Material
Download TIFF Image (370.2 KB)Acknowledgments
The author thanks NewCore BiodataStudio in Shanghai for sequencing date analysis; Jing Sun, Yan Zhang, Yahong Li, and Qing Wang for important clinical and scientific suggestions. The study was supported by the talent project of Tianjin medical university eye hospital. No honorarium, grant, or other forms of payment were given to anyone to produce the manuscript. Each author listed on the manuscript has seen and approved the submission of this version of the manuscript and takes full responsibility for the manuscript.
Disclosure statement
The authors declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Holden BA, Fricke TR, Wilson DA, et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050. Ophthalmology. 2016;123(5):1036–1042.
- Wong TY, Ferreira A, Hughes R, et al. Epidemiology and disease burden of pathologic myopia and myopic choroidal neovascularization: an evidence-based systematic review. Am J Ophthalmol. 2014;157(1):9–25 e12.
- Wong YL, Saw SM. Epidemiology of pathologic myopia in asia and worldwide. Asia Pac J Ophthalmol (Phila). 2016;5(6):394–402.
- Bruce A, Re: Holden, et al. Global prevalence of myopia and high myopia and temporal trends from 2000 through 2050 (Ophthalmology 2016;123:1036-1042). Ophthalmology. 2017;124(3):e24–e25.
- Saw SM, Gazzard G, Shih-Yen EC, et al. Myopia and associated pathological complications. Ophthalmic Physiol Opt. 2005;25(5):381–391.
- Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5ʹ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–386.
- Batista PJ, Molinie B, Wang J, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15(6):707–719.
- Zhao BS, He C. Fate by RNA methylation: m6A steers stem cell pluripotency. Genome Biol. 2015;16:43.
- Shen L, Liang Z, Gu X, et al. N(6)-methyladenosine RNA modification regulates shoot stem cell fate in arabidopsis. Dev Cell. 2016;38(2):186–200.
- Lichinchi G, Gao S, Saletore Y, et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. 2016;1:16011.
- Lin S, Choe J, Du P, et al. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345.
- Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047–2056.
- Wang Y, Mao J, Wang X, et al. Genome-wide screening of altered m6A-tagged transcript profiles in the hippocampus after traumatic brain injury in mice. Epigenomics. 2019;11(7):805–819.
- Wang Y, Zeng L, Liang C, et al. Integrated analysis of transcriptome-wide m(6)A methylome of osteosarcoma stem cells enriched by chemotherapy. Epigenomics. 2019;11(15):1693–1715.
- Wu Q, Yuan X, Han R, et al. Epitranscriptomic mechanisms of N6-methyladenosine methylation regulating mammalian hypertension development by determined spontaneously hypertensive rats pericytes. Epigenomics. 2019;11(12):1359–1370.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12.
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360.
- Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511.
- Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for gene ontology. R Package Version. 2010;2:2010.
- Tian L, Greenberg SA, Kong SW, et al. Discovering statistically significant pathways in expression profiling studies. Proc Nat Acad Sci. 2005;102(38):13544–13549.
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192.
- Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS. Genome Biol. 2008;9(9):R137.
- Shen L, Shao NY, Liu X, et al. diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PloS One. 2013;8(6):e65598.
- TL B. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27(12):1653–1659.
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206.
- Wei CM, Gershowitz A, Moss B. 5ʹ-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry. 1976;15(2):397–401.
- Schibler U, Kelley DE, Perry RP. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol. 1977;115(4):695–714.
- Luo GZ, MacQueen A, Zheng G, et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat Commun. 2014;5:5630.
- Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120.
- Niu Y, Zhao X, Wu YS, et al. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013;11(1):8–17.
- Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3ʹ UTRs and near stop codons. Cell. 2012;149(7):1635–1646.
- Csepany T, Lin A, Baldick CJ Jr., et al. Sequence specificity of mRNA N6-adenosine methyltransferase. J Biol Chem. 1990;265(33):20117–20122.
- Harper JE, Miceli SM, Roberts RJ, et al. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990;18(19):5735–5741.
- Wei CM, Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16(8):1672–1676.
- Wan Y, Tang K, Zhang D, et al. Transcriptome-wide high-throughput deep m(6)A-seq reveals unique differential m(6)A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 2015;16:272.
- Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–543.
- Jia R, Chai P, Wang S, et al. m(6)A modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol Cancer. 2019;18(1):161.
- Luo G, Xu W, Zhao Y, et al. RNA m(6) A methylation regulates uveal melanoma cell proliferation, migration, and invasion by targeting c-Met. J Cell Physiol. 2020;235:7107–7119.
- Yang J, Liu J, Zhao S, et al. (6)-Methyladenosine METTL3 modulates the proliferation and apoptosis of lens epithelial cells in diabetic cataract. Mol Ther Nucleic Acids. 2020;20:111–116.
- Fu Y, Dominissini D, Rechavi G, et al. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15(5):293–306.
- Yue Y, Liu J, He C, et al. methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29(13):1343–1355.
- Xu C, Wang X, Liu K, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–929.
- Wang X, Zhao BS, Roundtree IA, et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161(6):1388–1399.
- Scheckel C, Drapeau E, Frias MA, et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. eLife. 2016;5:e10421.
- Loiselle JJ, Sutherland LC. RBM10: harmful or helpful-many factors to consider. J Cell Biochem. 2018;119(5):3809–3818.
- Martinez-Arribas F, Agudo D, Pollan M, et al. Positive correlation between the expression of X-chromosome RBM genes (RBMX, RBM3, RBM10) and the proapoptotic Bax gene in human breast cancer. J Cell Biochem. 2006;97(6):1275–1282.
- Amanchy R, Zhong J, Molina H, et al. Identification of c-Src tyrosine kinase substrates using mass spectrometry and peptide microarrays. J Proteome Res. 2008;7(9):3900–3910.
- Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189.
- Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95.
- Wang Y, Li Y, Toth JI, et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191–198.
- Wang X, Sun B, Jiang Q, et al. mRNA m(6)A plays opposite role in regulating UCP2 and PNPLA2 protein expression in adipocytes. Int J Obes (Lond). 2018;42(11):1912–1924.
- Lee CG, Hartl D, Lee GR, et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med. 2009;206(5):1149–1166.
- Shao R, Taylor SL, Oh DS, et al. Vascular heterogeneity and targeting: the role of YKL-40 in glioblastoma vascularization. Oncotarget. 2015;6(38):40507–40518.
- Kang JY, Jo MR, Kang HH, et al. Long-term azithromycin ameliorates not only airway inflammation but also remodeling in a murine model of chronic asthma. Pulm Pharmacol Ther. 2016;36:37–45.
- Yan L, Deng Y, Zhou J, et al. China Hep BRFARG. Serum YKL-40 as a biomarker for liver fibrosis in chronic hepatitis B patients with normal and mildly elevated ALT. Infection. 2018;46(3):385–393.
- Zhou LX, Shao L, Xu L, et al. The relationship between scleral staphyloma and choroidal thinning in highly myopic eyes: the Beijing Eye Study. Sci Rep. 2017;7(1):9825.