
ABSTRACT
Accumulating evidence suggests that individuals exposed to victimization at key developmental stages may have different epigenetic fingerprints compared to those exposed to no/minimal stressful events, however results are inconclusive. This study aimed to strengthen causal inference regarding the impact of adolescent victimization on the epigenome by controlling for genetic variation, age, gender, and shared environmental exposures. We conducted longitudinal epigenome-wide association analyses (EWAS) on DNA methylation (DNAm) profiles of 118 monozygotic (MZ) twin pairs from the Environmental Risk study with and without severe adolescent victimization generated using buccal DNA collected at ages 5, 10 and 18, and the Illumina EPIC array. Additionally, we performed cross-sectional EWAS on age-18 blood and buccal DNA from the same individuals to elucidate tissue-specific signatures of severe adolescent victimization. Our analyses identified 20 suggestive differentially methylated positions (DMPs) (P < 5e-05), with altered DNAm trajectories between ages 10–18 associated with severe adolescent victimization (∆Beta range = −5.5%−5.3%). Age-18 cross-sectional analyses revealed 72 blood (∆Beta range = −2.2%−3.4%) and 42 buccal (∆Beta range = −3.6%−4.6%) suggestive severe adolescent victimization-associated DMPs, with some evidence of convergent signals between these two tissue types. Downstream regional analysis identified significant differentially methylated regions (DMRs) in LGR6 and ANK3 (Šidák P = 5e-09 and 4.07e-06), and one upstream of CCL27 (Šidák P = 2.80e-06) in age-18 blood and buccal EWAS, respectively. Our study represents the first longitudinal MZ twin analysis of DNAm and severe adolescent victimization, providing initial evidence for altered DNA methylomic signatures in individuals exposed to adolescent victimization.
Introduction
Exposure to stress during childhood and adolescence is detrimental to adult health and findings from a number of studies have linked early-life stress with a range of psychiatric and physical disorders that persist into adulthood [Citation1–3]. A recent retrospective survey conducted by the World Health Organization reported that nearly 40% of adults experienced some form of severe stress during childhood and/or adolescence [Citation4]. However, there is evidence of high inter- and intra-individual variability and adaptability in the stress response system resulting from a complex interaction between multiple genes and the social environment [Citation5,Citation6]. Although there is accumulating evidence suggesting that exposure to early-life stress, including victimization, leads to adverse outcomes in later life [Citation7,Citation8], the potential mechanisms underlying the ‘biological embedding’ of these psychosocial experiences are less well understood. One possibility is that environmental conditions could affect or interact with genes through epigenetic mechanisms, including DNA methylation (DNAm), which may mediate long-term effects on health. DNA methylation can alter the way in which genes are expressed without inducing changes in the actual sequence of the genes, thereby, having functional consequences [Citation9].
Epigenetic processes are dynamic and can fluctuate across the lifespan in response to genetic and environmental influences, especially during key developmental periods such as early childhood and adolescence [Citation10]. However, some epigenetic patterns especially those required for cell-lineage classification may be retained as a form of epigenetic memory [Citation11]. There has been a steady rise in studies linking victimization exposure (such as physical and sexual abuse, neglect, and bullying by peers) to changes in DNAm [Citation12–15] but definitive evidence is lacking due to diverse study populations including different ages and ethnic backgrounds, inconsistent methodology and non-overlapping results. Many of the studies conducted to date adopted a cross-sectional, case-control design which do not account for changes in DNA methylation over time, nor the underlying genetic differences, known to affect liability to stress reactivity and its interaction with victimization exposure as well as epigenetic processes [Citation14,Citation16,Citation17]. Some of these findings might also be confounded by potential recall and recruitment bias, e.g. the use of adult retrospective reports of stress or trauma, unusual clinical groups such as suicide victims or institutionalized children, or relatively small sample sizes [Citation18–22]. Moreover, childhood victimization has been the focus of the majority of these studies with very few investigating the association between victimization in adolescence, a potentially key sensitive window for long-term physiological and behavior changes [Citation23,Citation24] in which victimization exposures peak [Citation25] and altered epigenetic profiles have been reported [Citation14,Citation26,Citation27]. Adolescence is also an important developmental phase to focus upon because the majority of individuals who experience severe mental health problems develop them during this period [Citation28] and many of these have been associated with exposure to victimization in adolescence [Citation29–31].
To this end, we performed a genetically-informed epigenome-wide association study (EWAS) to explore the impact of severe victimization during adolescence on the epigenome by combining the monozygotic (MZ) twin design with a longitudinal approach [Citation32,Citation33]. Also, to isolate epigenetic patterns associated with adolescent victimization we purposely selected twins where neither twin had been exposed to severe victimization during childhood (that is not exposed to severe physical, sexual or emotional abuse, bullying by peers, neglect, or domestic violence by age 12 years). In our main analyses, we conducted a longitudinal EWAS using methylation data from buccal DNA collected from the same individual before and after severe adolescent victimization exposure and compared these profiles to those of the unexposed twins. This enabled us to explore the longitudinal epigenetic trajectories associated with severe adolescent victimization and minimized the potentially confounding effects of genetic variation, age, sex, and shared environmental exposures that are common limitations of previous epigenetic studies. We also performed parallel EWAS in the blood and buccal samples obtained from the same individuals at age 18 and explored the potential tissue-specific epigenetic signatures associated with severe adolescent victimization. Finally, taking advantage of the discordant MZ twin design, we conducted an exploratory analysis of within-twin-pair methylation changes where one twin in each pair had been exposed to severe adolescent victimization, while the other had not, to more stringently control for unmeasured shared environmental and genetic factors.
Materials and methods
Study cohort
Participants were members of the Environmental Risk (E-Risk) Longitudinal Twin Study described in detail previously [Citation34] and in the Supplementary Methods. Briefly, the E-Risk study tracks the development of a 1994–1995 birth cohort of 2,232 British children. The study sample was constructed in 1999 and 2000, when 1,116 families (93% of those eligible) with same-sex 5-year-old twins participated in home-visit assessments. This sample comprised 56% monozygotic (MZ) and 44% dizygotic (DZ) twin pairs, and sex was evenly distributed within zygosity (49% male). Home visits were conducted when participants were aged 5, 7, 10, 12, and 18 years with 93% retention. There were no differences between those who did and did not take part at age 18 in terms of socioeconomic status (SES) assessed when the cohort was initially defined (χ2 = 0.86, p = 0.65), age-5 IQ scores (t = 0.98, p = 0.33), or age-5 internalizing or externalizing behavior problems (t = 0.40, p = 0.69 and t = 0.41, p = 0.68, respectively). The Joint South London and Maudsley and the Institute of Psychiatry Research Ethics Committee approved each phase of the study. Parents gave informed consent, and participants gave assent at ages 5–12 and informed consent at age 18.
Victimization exposure
Childhood and adolescent victimization experiences in this cohort have been described previously [Citation35,Citation36] and are summarized briefly here. Full details are provided in the Supplementary Methods.
Childhood victimization (0-12 years)
Exposure to childhood victimization since birth was assessed repeatedly when the children were 5, 7, 10, and 12 years old, including exposure to violence between the mother and her partner, frequent bullying by peers, physical maltreatment by an adult, sexual abuse, emotional abuse and neglect, and physical neglect. Each exposure across childhood was coded on a 3-point scale (0 = no exposure, 1 = probable/less severe exposure, 2 = definite/severe exposure). All our study participants were selected for having no severe victimization exposure by age 12.
Adolescent victimization (12-18 years)
At age 18, each twin was interviewed separately about exposure to a range of victimization experiences between ages 12 and 18 using the Juvenile Victimization Questionnaire (JVQ) [Citation37,Citation38] adapted as a clinical interview [Citation36]. Seven forms of victimization were assessed: maltreatment, neglect, sexual victimization, family violence, peer/sibling victimization, cyber-victimization, and crime victimization. Each of these was rated as 0 (no exposure), 1 (some exposure), or 2 (severe exposure) by trained raters based on the descriptions of the experiences provided by participants and using the coding system of the Childhood Experience of Care and Abuse interview manual [Citation39]. Only those with a score of 2 for at least one type of victimization were considered to have been exposed to severe adolescent victimization.
Three groups of MZ twin pairs were selected for the current epigenetic study: a) Group 1: discordant MZ twin pairs where only one twin in the pair had reported severe adolescent victimization (N = 62), b) Group 2: concordant unexposed MZ twin pairs where both twins had reported no severe adolescent victimization (N = 28), and c) Group 3: concordant exposed MZ twin pairs where both twins had reported severe adolescent victimization (N = 28) (Supplementary Table 1).
Genome-wide DNA methylation analysis
Buccal samples were collected from participants at ages 5, 10 and 18 and whole blood was collected at age 18. Genomic DNA was extracted using standard protocols [Citation40–42]. 500ng of buccal and blood DNA was treated with sodium bisulphite using the EZ96 DNA Methylation kit (Zymo Research, Irvine, California) following the manufacturer’s standard protocol. Repeated samples from twins and their co-twin, including blood and buccal DNA, were processed on the same 96-well plate, and twin pairs belonging to different categories were randomized to minimize potential batch effects. DNAm was assessed using the Illumina Infinium HumanMethylationEPIC BeadChip kit (Illumina, Inc., San Diego, California) and quantified on an Illumina HiScan System (Illumina, Inc.). The level of methylation is expressed as a ‘beta’ value (β-value), ranging from 0 (no cytosine methylation) to 1 (complete cytosine methylation).
All data pre-processing and downstream statistical analyses were performed using R version 3.4.3 [Citation43]. Data quality control (QC) of the methylation profiles is detailed in Supplementary Methods. After stringent QC, the final dataset comprised 736/944 (80%) samples (see details in Supplementary Table 1) and 695,834 probes for downstream statistical analyses. Cell-type composition was estimated using the Houseman algorithm [Citation44] in the blood samples and EPiDISH package [Citation45] in the buccal samples to adjust for the potential differential cellular heterogeneity. Age-18 smoking pack-year data were used as covariates in all relevant analyses. QQplots and regional Manhattan plots were generated using the R packages qqman [Citation46] and ggplot2. The dataset is accessible from the Gene Expression Omnibus database (accession number: GSE154566).
Statistical analyses
In this study, we investigated the possible associations between severe adolescent victimization exposure and differential DNA methylation using two statistical models, an unpaired main analysis where all individuals (groups 1, 2 and 3) were treated as singletons to maximize power while adjusting for their relatedness structure in the dataset, and a paired secondary analysis where we studied the twin intra-pair differences (see Supplementary Figure 1).
Main analyses
Longitudinal EWAS using buccal DNA
Methylation β-values from all three time-points (ages 5, 10 and 18) from all the MZ twins in each of the groups (groups 1, 2 and 3, N = 501) (Supplementary Table 1, Supplementary Figure 1a) were treated as singletons whilst adjusting for their relatedness structure in the dataset and modelled over time using linear regression with clustered robust standard errors to account for the non-independence of twin observations [Citation47]. The model was fitted individually for each CpG, with severe adolescent victimization as the exposure of interest and DNAm as the outcome with age, gender, cell-type proportions and smoking status (smoking pack-years at age 18) as covariates. An interaction term for age and severe adolescent victimization was included to dissect the specific effect of exposure on methylation change during childhood (5–10 years) or adolescence (10–18 years).
A simplified version of the model formula is:
DNA methylation ~ Victimization exposure + sex + age + smoking status + cell types + victimization exposure * age, cluster = FamilyID
We used an EPIC array experiment-wide significance threshold of 9e-08 [Citation48] and a suggestive significance P-value threshold of P < 5e-05 to identify DMPs associated with severe adolescent victimization (i.e. between ages 10 and 18) in this unpaired analysis.
Cross-sectional EWAS using age-18 blood and buccal DNA
We also performed parallel EWASs to identify potential severe adolescent victimization-associated DNAm variation in age-18 blood and buccal samples using all twins in groups 1, 2 and 3 treated as singletons (Supplementary Figure 1a) whilst adjusting for their relatedness structure in the dataset using linear regression with clustered robust standard errors [Citation47]. Both the models included gender, cell-type proportions and smoking pack years as covariates. The formula is as described here:
DNA methylation ~ Victimization exposure + sex + smoking status + cell types, cluster = FamilyID
Similar to the longitudinal analysis, a suggestive P-value threshold of P < 5e–05 and an EPIC array significance threshold of P < 9e–08 were used to identify potential DMPs associated with severe adolescent victimization. We also performed additional exploratory analyses to check for the robustness of the age-18 blood EPIC array data using matched 450 K array data (see Supplementary Methods).
Exploratory analyses
We capitalized on the availability of twin level data within our study to perform an exploratory paired analysis to identify longitudinal methylation change as a result of severe adolescent victimization by including EWAS data from only complete discordant MZ twin pairs at age 10 and 18 (i.e. group 1, n = 24) (Supplementary Figure 1b). The major advantage of this is that it allows us to fully control for genetic and unmeasured shared environmental influences. Briefly, intra-individual changes in buccal DNAm from ages 10 to 18 were calculated (longitudinal ∆β) and the difference in the longitudinal ∆β between the exposed twin and their unexposed co-twin was examined using a paired t-test. We used the established ranked magnitude-significance method [Citation49,Citation50] for the identification of differentially methylated probes. In brief, CpGs were ranked separately using paired t-test P-value (significance) and the magnitude of the difference in DNAm change (absolute ∆β) and a final ranked list was determined by adding the two ranks. For the cross-sectional analyses, paired t-tests were performed separately in the age-18 blood and buccal DNA samples for the discordant twin pairs and the top 10 DMPs were identified using the ranked magnitude-significance method described above. The specificity of the top 10 DMPs associated with severe adolescent victimization in the longitudinal and the cross-sectional discordant twin analyses was determined by examining the within-twin DNAm differences at these loci in [Citation1] concordant unexposed control MZ twins (both twins did not have exposure to severe adolescent victimization, group 2), and [Citation2] concordant exposed twins (both twins exposed to severe adolescent victimization, group 3). The group differences were assessed using a one-way analysis of variance (ANOVA) and post hoc pairwise comparisons (pairwise t test) were performed to identify which groups were significantly different from each other.
Differentially methylated regions analysis
We used the Python module Comb-p [Citation51] to identify DMRs grouping spatially correlated DMPs (seed P-value<1 × 10−4, minimum of three probes) at a maximum distance of 500bp for both the main and exploratory analyses. DMR P-values were corrected for multiple testing using the Šidák correction [Citation52] as implemented as default in Comb-p.
Gene ontology pathway analysis
Illumina UCSC gene annotation was used to create a test gene list from the DMPs (P ≤ 5e-5) in the longitudinal and cross-sectional EWASs separately. This was performed for the main and exploratory analyses results separately. Gene ontology and pathway analysis were performed using the missMethyl package [Citation53–55] which takes into account the variable number of EPIC probes associated with each gene. The KEGG pathways were also investigated using the missMethyl package to provide further insights into the relevant biological processes associated with the DMPs (P < 5e-05). Independent pathways with FDR <0.05 were considered to be associated with severe adolescent victimization.
Results
Longitudinal DNAm changes in MZ twins with differing exposures to adolescent victimization
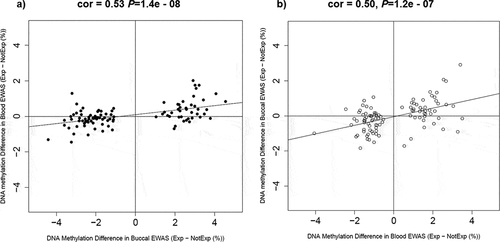
An overview of our study is illustrated in . Although none of the differentially methylated positions (DMPs) passed the stringent EPIC-array threshold (P < 9e–08) in our primary unpaired analysis, we identified 20 severe adolescent victimization-associated DMPs that passed the ‘discovery’ P-value threshold of P < 5e-5 (, Supplementary Figure 2). The trajectories for DNAm at the three top-ranked severe adolescent victimization-associated DMPs are detailed in –. The top-ranked probe cg02131853 (∆Beta = 3.43%, P = 1.23e-06), mapping upstream of TMEM156 gene, exhibited a differential trajectory of DNAm change from ages 10–18 between exposed and unexposed twins ().
Table 1. Top DMPs (P < 5e-05) associated with severe adolescent victimization in the unpaired analysis
Figure 1. An overview of the study design
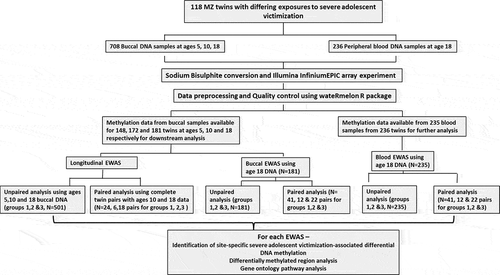
Figure 2. The epigenetic trajectories for the three top ranked differentially methylated probes (a-c) in the longitudinal epigenome-wide association study for severe adolescent victimization in exposed (solid line) and unexposed (dotted line) twins.
In our exploratory paired analysis, where we investigated the within twin-pair longitudinal ∆β change between ages 10 and 18 in the discordant MZ twin pairs (n = 24) using the ranked magnitude-significance method, we identified the cg09348925 probe as the most associated finding (∆Beta = 15.4%, P = 1.32e-05), located on chromosome 20 with the closest gene being a zinc finger protein gene ZNF217 (approx. 200 kb upstream) (, Supplementary Figure 3a). We next tested the specificity of the top 10 severe adolescent victimization-associated DMPs by comparing the average within-twin longitudinal DNA methylation differences with those from six age-matched concordant unexposed MZ twin pairs (where neither twin was exposed to severe adolescent victimization) and 18 concordant exposed MZ twin pairs (where both the twins were exposed to severe adolescent victimization). At two of the top-10 DMPs, the average within-twin ∆β was significantly larger in the discordant twins compared to the concordant exposed and concordant unexposed twins (see Supplementary Figure 3b) with post hoc pairwise comparisons indicating that average within-twin differences in DNA methylation are significantly larger at these top-ranked DMPs in the discordant twins compared to the twins concordant for exposure to severe adolescent victimization.
Table 2. Top ranked list of DMPs associated with severe adolescent victimization in the paired analysis of discordant MZ twin pairs
No differentially methylated regions (DMRs) were identified in our longitudinal unpaired and paired analyses. Downstream gene ontology (GO) and KEGG enrichment analysis on genes annotated to the severe adolescent victimization-associated DMPs (P < 5e-05) in the unpaired analyses identified significant enrichment of associated DMPs in KEGG pathways including lipid metabolism and inflammatory mediator regulation of TRP channels (Supplementary Table 2). In our KEGG analysis of the paired longitudinal EWAS results, 16 pathways were significantly associated (FDR<0.5) with severe adolescent victimization including linoleic acid metabolism and arachidonic acid metabolism pathways (Supplementary Table 3) common to the unpaired KEGG pathway results.
Site-specific DNAm differences in MZ twins with differing exposures to severe adolescent victimization in blood and buccal DNA at age 18
In our unpaired cross-sectional age-18 blood EWAS, we observed considerable variability in the DNAm at individual CpG sites within severe adolescent victimization-exposed and unexposed twins, although none of the DMPs survived multiple testing, in line with those reported by Marzi et al.[Citation14] in a related analysis of the full E-Risk cohort. Specifically, our age-18 blood dataset revealed 72 severe victimization-associated DMPs (P < 5e-5; , Supplementary Figure 4) annotated to 54 genes with effect sizes (mean methylation difference between exposed and unexposed groups) ranging from −2.2% to 3.4% (). The top ranked DMP cg21566892, which mapped to the intragenic region of the CPA6 gene encoding a metallocarboxypeptidase, was significantly hypomethylated (∆Beta = −1.6%, P = 4.16e-07) in severe victimization-exposed twins compared to unexposed twins (Supplementary Figure 5a).
In the exploratory paired age-18 blood analysis including DNAm data from 41 discordant twin pairs (complete pairs with age-18 blood and buccal data after QC), we identified the cg25412677 probe on Chr3q26.33 as the most associated finding (Beta = ∆3.3%, P = 4.80e-05) (, Supplementary Figure 6a). Of note, we did not observe any significant difference in smoking behavior between the exposed and unexposed twins within the twin pairs group discordant for severe adolescent victimization exposure (P = 0.573). Interestingly, we reported significant differences in the average within-twin methylation values for seven of the top 10 associated loci between the discordant twins and concordant exposed and unexposed twins (groups 1, 2 and 3, Supplementary Figure 6b), with three of these showing higher average within-twin methylation differences in the discordant twin group compared to both the concordant exposed and unexposed twins (Supplementary Figure 6b).
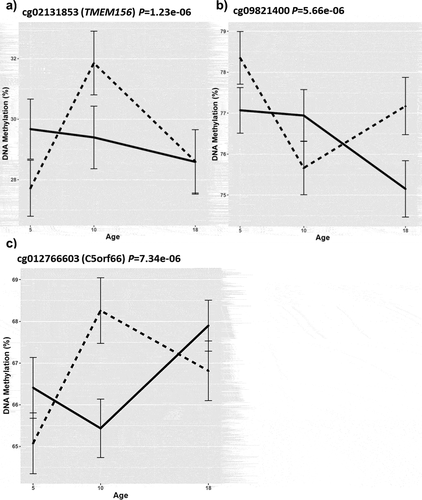
Using a regional approach, we identified two victimization-associated DMRs in LGR6 (Šidák P-value: P = 5e-09) and ANK3 (Šidák P-value: P = 4.07e-06) ( and 3b, Supplementary Table 4) in the unpaired EWAS. No DMRs were identified in our paired discordant twin analysis. Downstream pathway analysis of the unpaired and paired age-18 blood EWAS results did not reveal any enrichment of independent GO and KEGG pathways.
Figure 3. DNA methylation profiles of probes identified within the severe adolescent victimization-associated DMRs, including (a) LGR6 and (b) ANK3 in the age-18 blood, and c) CCL27 in the age-18 buccal epigenome-wide association study.
To check for the robustness of the age-18 blood EPIC results, we performed additional exploratory analyses on overlapping 450 K data on matched samples and observed some evidence of consistency in the directional effect of 33 out of 72 (P < 5e-05) severe adolescent victimization-associated probes common across the EPIC and 450 K arrays (Pbinomial = 0.01) (Supplementary Figure 7). However, it is worth noting that the individual probe correlations for the DNAm value across the two arrays were variable including that for probes reported in the current study with P < 5e-05 (21% sites with r > 0.5) (as detailed in Supplementary Table 5).
In the age-18 buccal unpaired EWAS, we identified 42 DMPs (mapped to 28 genes, P < 5e-5) with effect sizes ranging from −3.6% to 4.55% (, Supplementary Figure 8) that were associated with severe adolescent victimization. The top-ranked DMP, cg20000688 (Beta = 2.32%, P = 9.19e-07, Supplementary Figure 5b), was hypermethylated in twins exposed to severe adolescent victimization compared to the unexposed twins and mapped to a long noncoding RNA gene LINC01182. In the exploratory paired discordant twin analysis, the probe cg12971523 (∆Beta = −6.6%, P = 3.6e-05, , Supplementary Figure 9a) was the most associated finding. Of note, the second ranked probe cg24937727 (∆Beta = 5.9%, P = 3.9e-05) was also associated (P < 5e-05) in the unpaired analysis (∆Beta = 4.07%, P = 3.54e-05, ) and is located intragenic in a CpG island in RGL3 suggesting a robust methylation difference in the age-18 buccal tissue of the exposed and unexposed twins. At five of the 10 top-ranked DMPs, the average within-twin differences in DNA methylation were significantly different between the groups (Supplementary Figure 9b).
We identified a DMR associated with severe victimization upstream of the CCL27 gene (Šidák P-value: P = 2.80e-06, , Supplementary Table 4) in the age-18 buccal unpaired regional analysis. Pathway analysis did not identify any enrichment of GO and KEGG biological pathways in the unpaired analysis, however, for the paired age-18 buccal EWAS results, the GO analysis revealed homophilic cell adhesion via plasma membrane adhesion molecules pathway (Supplementary Table 6).
Severe adolescent victimization-associated methylomic differences are shared between peripheral tissues
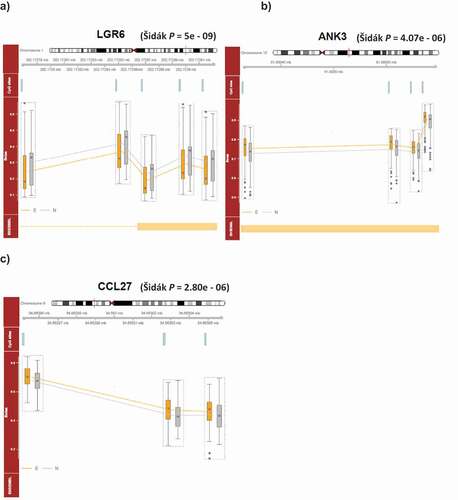
We next examined the extent to which severe adolescent victimization-associated DNAm differences are shared between tissue types (blood and buccal) using results from the unpaired analysis. Despite the distinct lists of the top 100 adolescent victimization-associated DMPs in the age-18 blood and age-18 buccal EWAS, there were positive correlations between the effect sizes of victimization-associated DMPs in the two datasets. Specifically, the effect sizes of the top 100 severe victimization-associated DMPs (i.e., the change in DNA methylation at the probe-level as a result of victimization, controlling for cell composition, smoking history and gender) in the buccal EWAS were moderately positively correlated with those of the same probes in the blood dataset (r = 0.53, P = 1.4e-08, ). Similarly, moderate cross-tissue positive correlations were present for the effect sizes of the top 100 DMPs in the blood EWAS analyses when compared to the buccal dataset (r = 0.50, P = 1.2e-07, ).
Figure 4. The effect sizes of the top 100 severe adolescent victimization-associated DMPs in (a) age-18 buccal, and (b) age-18 blood samples showed strong significant positive correlations with the severe adolescent victimization-associated effect sizes of the same probes from the other peripheral tissue type.
Discussion
To our knowledge, this represents the first comprehensive analysis of DNAm in relation to severe adolescent victimization that utilized a combined longitudinal, MZ twin discordance and genome-wide approach, also interrogating the potential tissue-specific epigenetic signatures associated with severe adolescent victimization using DNA collected from the same individual. In this study we performed an EWAS of severe adolescent victimization using two different statistical approaches, i.e. unpaired and paired linear regression (the former including clustered robust standard errors to account for the non-independence of twin observations). The unpaired method allowed us to correct for the effects of smoking, cell-types and gender on individual DNA methylation levels along with the inclusion of methylation data from all individuals thereby maximizing the power in detecting differential methylation associated with victimization exposure. The exploratory paired analyses explored the within twin-pair differences using the MZ twins discordant for severe adolescent victimization exposure, allowing us to control for genetic and unmeasured shared environmental influences, and we then investigated the specificity of the associated loci in concordant victimization-exposed and concordant unexposed twin pairs.
We report nominally-significant (P < 5e-5) altered epigenetic longitudinal trajectories associated with severe adolescent victimization at numerous CpG sites from our unpaired analysis, in genomic regions associated with stress response pathways including TMEM67 and HERPUD1 [Citation56,Citation57]. Our parallel cross-sectional unpaired EWASs in blood and buccal samples obtained from the same individuals at age 18 reported a non-overlapping list of nominally-significant (P < 5e-5) severe adolescent victimization-associated DMPs with some evidence of convergent signals between these two peripheral tissue types. Our exploratory paired analyses identified a DMP, cg12662887, common to the longitudinal and cross-sectional analyses. Future replication of this locus is required in independent samples. Notably, a CpG site in RGL3 was found to be significantly differentially methylated in the age-18 buccal samples in both our paired and unpaired analyses, suggesting a robust methylation change at this locus as a result of exposure to severe adolescent victimization. Interestingly, a DMR containing this CpG (Chr 19: 11,517,079–11,517,436) has previously been associated with alcohol intake in leukocyte DNA from women participating in the European Prospective Investigation into Cancer and Nutrition (EPIC) study [Citation58]. Another study investigating the effect of prenatal alcohol exposure on DNAm in buccal DNA from children with foetal alcohol spectrum disorder reported association with the same DMR [Citation59]. Given that victimized adolescents are more likely to consume alcohol [Citation60], it is plausible that our observed methylation change in RGL3 may be a reflection of this and future studies should further explore this link.
Although there has been a recent expansion in the literature documenting DNAm variation with early-life adversity specifically in childhood [Citation13,Citation20,Citation61], there is a paucity of studies in this field focusing on victimization during adolescence, with a bigger gap in the area of longitudinal research. A recent cross-sectional EWAS using whole blood in the complete E-Risk cohort of MZ and DZ twins reported limited evidence for an association between DNAm and several forms of early-life victimization [Citation14]. We did not identify any Bonferroni-corrected significant associations between DNAm changes and severe adolescent victimization in our age-18 blood EWAS mirroring the results from the previous study, however, there are also fundamental differences between these studies. Firstly, our use of the EPIC array compared to the 450 K array increased the coverage of CpG sites across the genome nearly two-fold, and secondly, only individuals free of any severe childhood victimization but who experienced severe adolescent victimization were included in the current study, and we focused only on MZ twins. Nevertheless, we examined methylation data available for the overlapping probes on the EPIC and 450 K arrays in matched samples from the two studies (see Supplementary Table 5). Despite observing some consistency in the direction of the associations between victimization and DNAm in age-18 blood when using the 450 K and the EPIC arrays (Supplementary Figure 7), the degree of overlap appears inconclusive and thus does not provide strong support for this part of the findings. The overall correlation between the two arrays was high (r = 0.99), although individual site correlations were variable including that for probes reported in the current study with P < 5e-05 (21% sites with r > 0.5), which is very similar to that reported in previous studies comparing the two arrays [Citation62,Citation63]. Therefore, this aspect of our results should be interpreted with caution and future studies using only data generated from one type of array might have limited replicability and generalizability.
Our regional analysis (unpaired approach) identified DMRs upstream of CCL27 in the age-18 buccal EWAS and in the genes LGR6 and ANK3 in the age-18 blood EWAS. The 135bp DMR in ANK3 was consistently hypermethylated in twins exposed to severe adolescent victimization. ANK3 is a scaffolding protein and genetic variants annotated to this gene are associated with various psychiatric disorders including schizophrenia, bipolar disorder, and autism [Citation64–66]. A recent cross-species combined methylome analysis performed in different tissues and time-points in rats, non-human primates and humans, all characterized by early-life stress, revealed consistent hypermethylation in ANK3 in the stressed groups across all the conditions and species [Citation67]. Findings from our study provide further support for a potentially important role of altered ANK3 DNAm in relation to stress exposure. It is interesting to note that some of the genes identified in our differential methylation analysis are expressed in both salivary glands and leukocytes, including RGL3, ANK3 and CCL27, raising the possibility of gum infection in our twins, despite adjusting for cell-type composition in our main analyses. Future studies that contain adolescent victimization exposure and dental records could explore this potentially interesting association further.
This study has several strengths. Firstly, our longitudinal design allowed us to ascertain the longitudinal epigenetic trajectories associated with severe adolescent victimization by comparing the DNA methylome of individuals before and after the exposure experience. Secondly, our MZ twin design allowed us to ascertain a purer effect of adolescent victimization on the epigenome controlling for potentially important confounders in epigenetic studies such as genetic variation, age, gender, and shared environmental exposure effects. Thirdly, known important confounders in epigenetic studies including smoking [Citation68] and cell type composition in blood and buccal cells [Citation69,Citation70] were controlled for in our analyses, thereby, disentangling their mediating effects on DNAm. Methylation at AHRR, especially at cg05575921, in blood has been consistently reported to be inversely associated with cigarette smoking [Citation71,Citation72]. In the current study, one of the severe adolescent victimization-associated DMPs in age-18 blood data was located in the first intron of AHRR (cg09634134, ∆Beta = −0.89%, P = 1.39E-05), highlighting the potential intricate relationship between smoking behaviours and severe stress exposures as victimized adolescents are more likely to smoke [Citation14,Citation73]. It is also possible that unmeasured confounders such as smoking intensity, duration, passive smoking or air pollution [Citation74] could explain some of the effects at this locus. We used pack years information to control for smoking for consistency in all our blood and buccal analyses rather than a smoking score, the latter being derived from DNA methylation values of smoking-associated CpG sites measured in blood samples (and thus not necessarily applicable to buccal samples). Lastly, our study sample came from a cohort that represents the full range of socioeconomic conditions in Great Britain and had 93% retention over 18 years, thereby, minimizing ascertainment and attrition bias.
Nonetheless, the results from our study should be considered in light of certain limitations. We were unable to explore associations between the DNA methylome and specific types of victimization due to the modest sample size of each subgroup. It is possible that subtype-specific analyses would yield further insights given recent findings of associations between altered DNA methylome and exposure to sexual and physical abuse [Citation75,Citation76]. Also, DNAm was quantified using the Illumina EPIC array; although this is a robust, highly reliable, and currently the best high-throughput platform with content spanning regulatory regions associated with the majority of known annotated genes, it interrogates DNAm at a relatively small proportion of sites across the whole genome. In this study, genome-wide DNAm profiling was performed on DNA extracted from buccal cells collected at ages 5, 10 and 18 and whole blood collected at age 18. There is no archived collection of longitudinal brain samples from twins discordant for severe adolescent victimization and no techniques currently available to explore DNAm in the brains of live individuals. Finally, there is increasing awareness of the importance of 5-hydroxymethyl cytosine (5-hmC) as an epigenetic marker [Citation77], although this modification cannot be distinguished from DNAm using standard bisulphite-based approaches. It is plausible that many of the adolescent victimization-associated differences identified in this study are confounded by modifications other than DNAm, however, it is important to note that markers such as 5-hmC are known to be highly expressed in brain tissues but at a much lower level in blood cells.
In summary, this is the first systematic longitudinal MZ twin discordant study to examine the association between genome-wide DNAm in severe adolescent victimization, providing preliminary evidence for altered DNA methylomic signatures in individuals exposed to severe victimization during adolescence, a key stage of development and a crucial period for the onset of psychiatric disorders. Follow-up studies are needed to explicitly test whether the severe adolescent victimization-associated DMPs identified in the present study associate with psychopathological outcomes and, if so, whether they may mediate the influence of severe adolescent victimization on later mental health.
Supplemental Material
Download Zip (2 MB)Acknowledgments
We are grateful to the study mothers and fathers, the twins, and the twins’ teachers for their participation. Our thanks to Professors Terrie Moffitt and Avshalom Caspi who founded the E-Risk study, members of the E-Risk team for their dedication, hard work, and insights, the Nuffield Foundation, the Avielle Foundation, and CACI, Inc.
This work was supported by a joint UK Economic and Social Research Council (ESRC) and Biotechnology and Biological Sciences Research Council (BBSRC) grant [ES/N000277/1] to CCYW. The E-Risk Study is supported by the Medical Research Council [G1002190]. This work was also supported by the National Institute of Child Health and Human Development [HD077482], the Jacobs Foundation, and an MQ Fellows Award [MQ14F40] to HLF. AD is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at the South London and Maudsley NHS Foundation Trust and King’s College London. JRS was supported by a grant from the Medical Research Council [MR/M025020/1]. LA is the Mental Health Leadership Fellow for the UK ESRC. HLF receives salary support from the ESRC Centre for Society and Mental Health at King’s College London [ES/S012567/1]. The views expressed are those of the authors and not necessarily those of the NHS, NIHR, Department of Health and Social Care, the ESRC or King’s College London.
Disclosure statement
The authors have no conflicts of interest to declare.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Danese A, McEwen BS. Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav. 2012;106:29–39.
- Dinkler L, Lundstrom S, Gajwani R, et al. Maltreatment-associated neurodevelopmental disorders: a co-twin control analysis. J Child Psychol Psychiatry. 2017;58:691–701.
- Shonkoff JP, Garner AS, Siegel, B.S., et al. The lifelong effects of early childhood adversity and toxic stress. Pediatrics. 2012;129:e232.
- Kessler RC, McLaughlin KA, Green JG, et al. Childhood adversities and adult psychopathology in the WHO World Mental Health Surveys. Br J Psychiatry. 2010;197:378–385.
- Agorastos A, Pervanidou P, Chrousos GP, et al. Early life stress and trauma: developmental neuroendocrine aspects of prolonged stress system dysregulation. Hormones (Athens). 2018;17:507–520.
- Schneiderman N, Ironson G, Siegel SD. Stress and health: psychological, behavioral, and biological determinants. Annu Rev Clin Psychol. 2005;1:607–628.
- Lupien SJ, McEwen BS, Gunnar MR, et al. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445.
- Miller GE, Chen E, Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol Bull. 2011;137:959–997.
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254.
- Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol. 2014;2:49.
- Kim M, Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017;49:e322.
- Barker ED, Walton E, Cecil CAM. Annual Research Review: DNA methylation as a mediator in the association between risk exposure and child and adolescent psychopathology. J Child Psychol Psychiatry. 2018;59:303–322.
- Labonte B, Suderman M, Maussion G, et al. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry. 2012;69:722–731.
- Marzi SJ, Sugden K, Arseneault L, et al. Analysis of DNA methylation in young people: limited evidence for an association between victimization stress and epigenetic variation in blood. Am J Psychiatry. 2018;175:517–529.
- Ouellet-Morin I, Wong CC, Danese A, et al. Increased serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response to stress in childhood: a longitudinal study of discordant monozygotic twins. Psychol Med. 2013;43:1813–1823.
- Caspi A, McClay J, Moffitt TE, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854.
- Zankert S, Bellingrath S, Wust S, et al. HPA axis responses to psychological challenge linking stress and disease: what do we know on sources of intra- and interindividual variability? Psychoneuroendocrinology. 2019;105:86–97.
- Esposito EA, Jones MJ, Doom JR, et al. Differential DNA methylation in peripheral blood mononuclear cells in adolescents exposed to significant early but not later childhood adversity. Dev Psychopathol. 2016;28:1385–1399.
- Lutz PE, Gross JA, Dhir SK, et al. Epigenetic regulation of the kappa opioid receptor by child abuse. Biol Psychiatry. 2018;84:751–761.
- Marinova Z, Maercker A, Grunblatt E, et al. A pilot investigation on DNA methylation modifications associated with complex posttraumatic symptoms in elderly traumatized in childhood. BMC Res Notes. 2017;10:752.
- Painter RC, Osmond C, Gluckman P, et al. Transgenerational efects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. 2008;115:1243–1249.
- Serpeloni F, Radtke K, de Assis SG, et al. Grandmaternal stress during pregnancy and DNA methylation of the third generation: an epigenome-wide association study. Transl Psychiatry. 2017;7:e1202.
- Gee DG, Casey BJ. The impact of developmental timing for stress and recovery. Neurobiol Stress. 2015;1:184–194.
- Yohn NL, Blendy JA. Adolescent chronic unpredictable stress exposure is a sensitive window for long-term changes in adult behavior in mice. Neuropsychopharmacol. 2017;42:1670–1678.
- Sickmund M, Puzzanchera C, eds. Juvenile Offenders and Victims: 2014 National Report. Pittsburgh, PA: National Center for Juvenile Justice; 2014.
- Dunn EC, Soare TW, Zhu Y, et al. Sensitive periods for the effect of childhood adversity on DNA methylation: results from a prospective, longitudinal study. Biol Psychiatry. 2019;85:838–849.
- Essex MJ, Boyce WT, Hertzman C, et al. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. 2013;84:58–75.
- Kim-Cohen J, Caspi A, Moffitt TE, et al. Prior juvenile diagnoses in adults with mental disorder: developmental follow-back of a prospective-longitudinal cohort. Arch Gen Psychiatry. 2003;60:709–717.
- Baldwin JR, Arseneault L, Caspi A, et al. Adolescent victimization and self-injurious thoughts and behaviors: a genetically sensitive cohort study. J Am Acad Child Adolesc Psychiatry. 2019;58:506–513.
- Newbury J, Arseneault L, Caspi A, et al. Cumulative effects of neighborhood social adversity and personal crime victimization on adolescent psychotic experiences. Schizophr Bull. 2018;44:348–358.
- Schaefer JD, Moffitt TE, Arseneault L, et al. Adolescent victimization and early-adult psychopathology: approaching causal inference using a longitudinal twin study to rule out noncausal explanations. Clin Psychol Sci. 2018;6:352–371.
- Sun YV. The influences of genetic and environmental factors on methylome-wide association studies for human diseases. Curr Genet Med Rep. 2014;2:261–270.
- Ng JW, Barrett LM, Wong A, et al. The role of longitudinal cohort studies in epigenetic epidemiology: challenges and opportunities. Genome Biol. 2012;13:246.
- Moffitt TE; E-Risk Study Team. Teen-aged mothers in contemporary Britain. J Child Psychol Psychiatry. 2002;43:727–742.
- Danese A, Moffitt TE, Arseneault L, et al. The origins of cognitive deficits in victimized children: implications for neuroscientists and clinicians. Am J Psychiatry. 2017;174:349–361.
- Fisher HL, Caspi A, Moffitt TE, et al. Measuring adolescents’ exposure to victimization: the Environmental Risk (E-Risk) Longitudinal Twin Study. Dev Psychopathol. 2015;27:1399–1416.
- Finkelhor D, Hamby SL, Turner H, et al. The Juvenile Victimization Questionnaire: 2nd Revision (JVQ-R2). Durham, NH: Crimes Against Children Research Center; 2011.
- Hamby SL, Finkelhor D, Ormrod R, et al. The Juvenile Victimization Questionnaire (JVQ): administration and Scoring Manual. Durham, NH: Crimes Against Children Research Centre; 2004.
- Bifulco A, Bernazzani O, Moran PM, et al. The childhood experience of care and abuse questionnaire (CECA.Q): validation in a community series. Br J Clin Psychol. 2005;44:563–581.
- Bowtell DD. Rapid isolation of eukaryotic DNA. Anal Biochem. 1987;162:463–465.
- Freeman B, Smith N, Curtis C, et al. DNA from buccal swabs recruited by mail: evaluation of storage effects on long-term stability and suitability for multiplex polymerase chain reaction genotyping. Behav Genet. 2003;33:67–72.
- Jeanpierre M. A rapid method for the purification of DNA from blood. Nucleic Acids Res. 1987;15:9611.
- R Core Team (2017) R: A Language and Environment for Statistical Computing.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
- Teschendorff AE, Breeze CE, Zheng SC, et al. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinformatics. 2017;18:105.
- Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J Open Source Softw. 2018;3:731.
- Staley JR, Suderman M, Simpkin AJ, et al. Longitudinal analysis strategies for modelling epigenetic trajectories. Int J Epidemiol. 2018;47:516–525.
- Mansell G, Gorrie-Stone TJ, Bao Y, et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 2019;20:366.
- Dempster EL, Pidsley R, Schalkwyk LC, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–4796.
- Dempster EL, Wong CC, Lester KJ, et al. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol Psychiatry. 2014;76:977–983.
- Pedersen BS, Schwartz DA, Yang IV, et al. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–2988.
- Sida´k Z. Rectangular confidence regions for the means of multivariate normal distributions. J Am Statist Assoc. 1967;62:626–633.
- Geeleher P, Hartnett L, Egan LJ, et al. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics. 2013;29:1851–1857.
- Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32:286–288.
- Young MD, Wakefield MJ, Smyth GK, et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14.
- Ho DV, Chan JY. Induction of Herpud1 expression by ER stress is regulated by Nrf1. FEBS Lett. 2015;589:615–620.
- Kokame K, Agarwala KL, Kato H, et al. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem. 2000;275:32846–32853.
- Perrier F, Viallon V, Ambatipudi S, et al. Association of leukocyte DNA methylation changes with dietary folate and alcohol intake in the EPIC study. Clin Epigenetics. 2019;11:57.
- Portales-Casamar E, Lussier AA, Jones MJ, et al. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin. 2016;9(1):25.
- Shepherd JP, Sutherland I, Newcombe RG. Relations between alcohol, violence and victimization in adolescence. J Adolesc. 2006;29:539–553.
- Houtepen LC, Hardy R, Maddock J, et al. Childhood adversity and DNA methylation in two population-based cohorts. Transl Psychiatry. 2018;8:266.
- Solomon O, MacIsaac J, Quach H, et al. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics. 2018;13:655–664.
- Sugden K, Hannon EJ, Arseneault L, et al. Patterns of reliability: assessing the reproducibility and integrity of DNA methylation measurement. Patterns. 2020;1:100014.
- Bi C, Wu J, Jiang T, et al. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat. 2012;33:1635–1638.
- Hughes T, Sonderby IE, Polushina T, et al. Elevated expression of a minor isoform of ANK3 is a risk factor for bipolar disorder. Transl Psychiatry. 2018;8:210.
- Guo X, Zhang Y, Du J, et al. Association analysis of ANK3 gene variants with schizophrenia in a northern Chinese Han population. Oncotarget. 2016;7:85888–85894.
- Luoni A, Massart R, Nieratschker V, et al. Ankyrin-3 as a molecular marker of early-life stress and vulnerability to psychiatric disorders. Transl Psychiatry. 2016;6:e943.
- Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–447.
- Ji H, Ehrlich LI, Seita J, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342.
- Liu H, Liu X, Zhang S, et al. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes. Nucleic Acids Res. 2015;44:75–94.
- Bojesen SE, Timpson N, Relton C, et al. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax. 2017;72:646–653.
- Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012;7:1331–1338.
- Norman RE, Byambaa M, De R, et al. The long-term health consequences of child physical abuse, emotional abuse, and neglect: a systematic review and meta-analysis. PLOS Med. 2012;9:e1001349.
- Tantoh DM, Lee KJ, Nfor ON, et al. Methylation at cg05575921 of a smoking-related gene (AHRR) in non-smoking Taiwanese adults residing in areas with different PM2.5 concentrations. Clin Epigenetics. 2019;11:69.
- Cecil CA, Smith RG, Walton E, et al. Epigenetic signatures of childhood abuse and neglect: implications for psychiatric vulnerability. J Psychiatr Res. 2016;83:184–194.
- Lawn RB, Anderson EL, Suderman M, et al. Psychosocial adversity and socioeconomic position during childhood and epigenetic age: analysis of two prospective cohort studies. Hum Mol Genet. 2018;27:1301–1308.
- Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2011;13:7–13.