
ABSTRACT
Ten-Eleven Translocation (TET) proteins convert 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) leading to a dynamic epigenetic state of DNA that can influence transcription and chromatin organization. While TET proteins interact with complexes involved in transcriptional repression and activation, the overall understanding of the molecular mechanisms involved in TET-mediated regulation of gene expression still remains limited. Here, we show that TET proteins interact with the chromatin remodelling protein lymphoid-specific helicase (LSH/HELLS) in vivo and in vitro. In mouse embryonic fibroblasts (MEFs) and embryonic stem cells (ESCs) knock out of Lsh leads to a significant reduction of 5-hydroxymethylation amount in the DNA. Whole genome sequencing of 5hmC in wild-type versus Lsh knock-out MEFs and ESCs showed that in absence of Lsh, some regions of the genome gain 5hmC while others lose it, with mild correlation with gene expression changes. We further show that differentially hydroxymethylated regions did not completely overlap with differentially methylated regions indicating that changes in 5hmC distribution upon Lsh knock-out are not a direct consequence of 5mC decrease. Altogether, our results suggest that LSH, which interacts with TET proteins, contributes to the regulation of 5hmC levels and distribution in MEFs and ESCs.
Introduction
5-methylcytosine (or 5mC) is an essential DNA modification in mammals. It plays a major role in a variety of biological and molecular processes during embryonic development including X-chromosome inactivation, genomic imprinting, transcription, chromatin organization and chromosome stability [Citation1–3]. 5mC is established by the DNA methyltransferases (DNMTs) [Citation1,Citation2,Citation4] and removed either catalytically by the Ten Eleven Translocation (TET) proteins or passively by dilution over cellular divisions in absence of maintenance [Citation5,Citation6].
TET proteins are 2-oxyglutarate and Fe(II)-dependent dioxygenases which are able to oxidize 5mC in 5-hydroxymethylcytosine (5hmC) and by further oxidation in 5-formylcytosine (5fC) and eventually in 5-carboxylcytosine (5caC) [Citation5,Citation6]. The discovery of TET enzymes has been instrumental in understanding the molecular basis of widespread changes in 5mC occurring during cell differentiation [Citation5–7]. Oxidation of 5mC can lead to its enzymatic excision and replacement by an unmethylated cytosine leading to DNA demethylation [Citation8,Citation9]. However, 5hmC can also persist as such and be detected at substantial levels in embryonic stem cells (ESCs) and neuronal cell types [Citation5–7,Citation10,Citation11]. In these cells, the mapping of 5hmC landscapes as well as mechanistic and genetic analyses have demonstrated important functions for 5hmC in gene expression, chromatin organization and cell fate decision [Citation12–16]. 5hmC is enriched in promoters, gene bodies, enhancers, and intergenic regions near genes, and increased levels of 5hmC often correlates with gene expression [Citation17,Citation18]. The identification and characterization of proteins involved in the establishment, removal and ‘reading’ of 5hmC is thus rising interest to get a deeper understanding of 5hmC function [Citation10,Citation19].
Various TETs interacting partners have been described, shedding some light on how TET proteins could act on 5hmC levels and distribution. Those include partners such as transcriptional factors/nuclear receptors (IDAX/CXXC4 [Citation20], NANOG [Citation21], PPARγ [Citation22], SP1/PU.1 [Citation23], EBF1 [Citation24], PRDM14 [Citation25], GADD45a [Citation26], NF-κB [Citation27] and ZSCAN4 [Citation28], to name a few), chromatin-associated proteins involved in transcriptional activation (OGT and SET1/COMPASS complex [Citation29]) or repression (SIN3A/HDACs, EZH2, NURD [Citation29,Citation30]) and the promyelocytic leukaemia (PML) protein [Citation31]. The depletion of these factors has various effects on TET enzymes functions, ranging from the regulation of their stability to their recruitment at specific loci or the regulation of their enzymatic activity. For instance, TET1 activity is regulated by the transcription factor NF-κB with an anti-correlation with the expression of genes related to immune response in various cancer cell lines [Citation27]. IDAX/CXXC4 depletion causes the caspase-dependent degradation of TET2 in differentiating mouse embryonic stem cells [Citation20]. Other studies indicated that TETs can also be regulated by microRNAs (miRNAs) [Citation32–36] or by post-translational modifications, including phosphorylation [Citation37–39] and O-GlcNAcylation [Citation40–42]. Finally, ascorbic acid (vitamin C) and retinoic acid have been shown to significantly enhance TET enzymatic activity. Ascorbic acid directly acts on TET activity as a specific enzymatic co-factor probably by reducing Fe(III) back to Fe(II) after catalysis [Citation43–47]. Retinoic acid, a key inducer of neuronal differentiation, indirectly acts on TET proteins activity by regulating their expression [Citation47,Citation48]. Altogether, these results shed lights on the regulatory mechanisms (post-translational modifications, miRNA network, small molecules) that impact TET proteins expression and/or activity. However, despite several studies addressing this issue, the precise mechanisms linking TET proteins to genomic patterns of 5hmC is not properly understood yet.
Chromatin-remodelling proteins play important molecular functions in transcription, DNA replication and DNA repair [Citation49]. The SNF2-like helicase LSH (also known as HELLS, SMARCA6 or PASG), initially identified as a factor required for lymphoid cells proliferation, belongs to the SNF2 family of chromatin-remodelling factors [Citation50,Citation51]. LSH employs the energy derived from ATP hydrolysis to disrupt histone/DNA interactions and allows the sliding of nucleosomes on the DNA in vitro [Citation52,Citation53]. This activity of LSH is crucial to regulate the accessibility of the DNA methyltransferases DNMT1, DNMT3A and DNMT3B to genomic sites [Citation53–61]. Lsh mutation or deletion often leads to a profound loss of global DNA methylation but can also lead to hypo- as well as hypermethylation at specific genomic loci, such as repetitive sequences and enhancers [Citation57,Citation59,Citation62–64]. In addition to reduced DNA methylation levels, Lsh-/- MEFs display an overall disorganization of chromatin, with alterations in nucleosome occupancy and histone modifications, such as histone H3 lysine 4 mono- and tri-methylation (H3K4me1 and H3K4me3), H3K27me3, H3K9me3 and histone variant macroH2A [Citation60,Citation65–67]. It is thus well established that LSH plays a major role in the regulation of chromatin organization and DNA methylation landscapes notably at enhancers and repetitive sequences [Citation55,Citation57]. Accordingly, it was reported that Lsh is essential for mouse development. Its deletion causes a lethal phenotype after birth with tissue-specific defects including skeletal defects, a smaller thymus and a barely detectable spleen [Citation51,Citation68,Citation69].
In this study, we investigated whether LSH contributes to the regulation of 5hmC landscapes. We showed that LSH and TETs interact in vivo in ESCs and in vitro. We found that knock-out (KO) of Lsh leads to a reduction in the global level of 5hmC in MEFs and ESCs. Genome-wide 5hmC studies in wild-type and Lsh KO MEFs and ESCs revealed that thousands of genomic regions gain or lose 5hmC. These changes in 5hmC occur mainly in gene bodies and at cis-regulatory elements of transcription, and in most cases mild changes in gene expression could be detected. Our data also showed that 5hmC modifications upon Lsh KO were not a direct consequence of changes in 5mC in these cells. Altogether, we identified the SNF2-like helicase LSH as a partner of TET enzymes and its lost leads to global and locus-specific effects on 5hmC levels.
Materials and methods
Cell culture and generation of Lsh KO ESCs
All cells were grown at 37°C in a humidified atmosphere of 5% CO2.
Wild-type and Lsh KO mouse embryonic fibroblasts (MEFs) were previously described [Citation56] and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 100 U/ml of penicillin/streptomycin.
V6.5 ESCs (kindly received from Dr K. Koh, KU Leven) [Citation70] were cultured as previously described [Citation71]. HEK293T/GP cells were cultured in DMEM, 10% FBS and penicillin/streptomycin as previously described [Citation29].
A modified knock-in strategy and allele design previously reported by Schnütgen et al [Citation72] were employed to generate the Lsh KO ESCs by sequential-targeted disruption of both Lsh alleles in E14 (129/Ola) ESCs (see Supplemental for a graphical illustration). We first integrated by homologous recombination a reversible stop cassette (SA-GFP-Neo) flanked by a set of LoxP and Frt sites into the third intron of the Lsh gene. The integrated stop cassette is predicted to generate a null Lsh allele, which we named Lshoff, producing a chimeric protein containing 72 amino acids of the LSH N-terminus, which lacks nuclear localization signal and any known function, fused to a GFP-Neomycin marker. The second Lsh allele was disrupted in one of the Lshoff/+ ES cell lines by targeted integration of the same stop cassette, but this time carrying a hygromycin resistance marker. The successful integration of both stop cassettes was confirmed by Southern and Western blots (Supplemental ). These Lshoff/off ESCs were named Lsh KO ESCs in this study. Wild-type and Lsh KO ESCs were expanded on feeders using regular ESC media (DMEM supplemented with 15% FBS, penicillin/streptomycin, non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine and 100 nM of β-mercaptoethanol) containing leukaemia inhibitory factor (LIF).
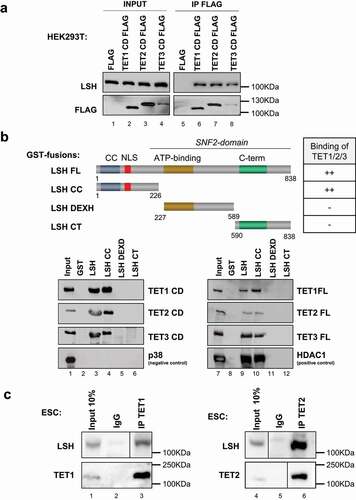
Figure 1. LSH is a TET interacting partner.
Transient transfection
TET Halo-tagged plasmids were already described [Citation29]. Flag-tagged plasmids encoding TET1, TET2 and TET3 catalytic domains (CD) were previously described [Citation6]. Polyethyleneimine (Euromedex) was used to transfect these different plasmids, or a combination of these, in HEK293T cells cultured in 100-mm diameter dishes as described previously [Citation73].
V6.5 ESCs were transfected with plasmids encoding murine Lsh shRNAs, empty pHyper vector (control) or non-relevant shRNA (NR) using Nucleofector 2B (Lonza). Sequences used for the design of shRNAs are KD1: CCGGCTAATCAGGGAGTTAAA, KD2: TCGAATGCTGCCCGAACTTAA, KD3: GGACACAGGATTAAGAATATG; and were cloned in pHYPER vector [Citation70]. Positively transfected cells were selected by growth in the presence of 1.5 µg/mL puromycin (Sigma).
Halo Tag (HT) mammalian pulldown assay
HT mammalian pulldown assays were performed as previously described [Citation29]. Briefly, HEK293T cells expressing HT-fusion proteins or HT-Ctrl were incubated in the mammalian lysis buffer (Promega) supplemented with Protease Inhibitor cocktail (Promega) and RQ1 RNase-Free DNase (Promega) for 10 min on ice.
The clarified lysate was incubated with HaloLink Resin (Promega) for 15 min at 22°C with rotation. The resin was then washed with wash buffer and protein interactors were eluted with SDS elution buffer. Affinity purified complexes were then analysed by nano-LC/MS/MS (MSBioworks, Michigan, USA; https://www.msbioworks.com/) and by Western blotting.
Immunoprecipitation assays
Whole-cell extracts were prepared in IPH lysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM EDTA, 0.5% NP40 supplemented with protease inhibitor cocktail (Roche). Immunoprecipitations were performed with anti-rabbit or anti-mouse IgG Dynabeads® (Life Technologies) overnight at 4°C. The primary antibodies used in these experiments were directed against the following: control immunoglobulin G (IgG) (rabbit sc-2027 and mouse sc-2025; Santa Cruz), FLAG M2 (F3165; Sigma), LSH (NB100–278; Novus), TET1 (09–872; Millipore) and TET2 (R1086–4; Abiocode). The immunoprecipitated complexes were eluted in Laemmli buffer and analysed by western blot.
GST pulldown assays
Full-length LSH, LSH coiled-coil or CC (amino-acids 1–226), LSH DEXH-box helicase domain containing the ATP domain (aa 227–589) and LSH C-terminal domain or CT (aa 590–838) domains cloned in the pGEX4T-1 vector were kindly provided by Dr U. Ziebold (Cancer Research, Berlin, Germany) [Citation74]. Recombinant GST-fused proteins were expressed in and purified from Escherichia coli BL21, as previously described [Citation74]. We performed in vitro transcription/translation using the TNT system (Promega) with pcDNA3 expression vectors for murine Flag-tagged TET catalytic domains (TET1, aa 1367–2039; TET2, aa 916–1921 and TET3, aa 697–1668) or full-length Halo-tagged human TET proteins. GST-pulldown assays were performed as previously described [Citation75].
Dot blot
Genomic DNA was extracted with the DNeasy blood and tissue kit (Qiagen), denatured for 10 min at 95°C, chilled directly on ice and 100 µg (ESC) or 200 µg (MEFs) spotted on a nylon membrane (GE Healthcare Hybond-N+). After UV-induced fixation of target nucleic acids (2 x 200 000 µJ/cm2 of UV), the membrane was blocked in PBS BSA 1% and incubated with anti-5hmC (39,769; Active Motif) or anti-ssDNA antibody (LS-C64821; LSBio). Goat Anti-Human IgG-HRP #2040–05 (Southern Biothech), Goat anti-Rabbit IgG-heavy and light chain #A120–101P (Bethyl) and are used as secondary antibody. Membranes revealed with SuperSignal West Dura Extended Duration Substrate (Thermo scientific) and images were quantified with ImageJ.
Analysis of global DNA 5mC and 5hmC levels by mass spectrometry (LC-MS/MS)
Analysis of global DNA 5mC and 5hmC levels by LC-MS/MS was carried out as described in Bachman et al [Citation11]. Briefly, 500 ng of genomic DNA was incubated with 5 units of DNA Degradase Plus (Zymo Research) at 37°C for 3 h. The resulting mixture of 2ʹ-deoxynucleosides was analysed on a Triple Quad 6500 mass spectrometer (AB Sciex) fitted with an Infinity 1290 LC system (Agilent) and an Acquity UPLC HSS T3 column (Waters), using a gradient of water and acetonitrile with 0.1% formic acid. External calibration was performed using synthetic standards, and for accurate quantification, all samples and standards were spiked with isotopically labelled nucleosides. 5mC and 5hmC levels are expressed as a percentage of total cytosines.
Identification of 5-hmC–enriched DNA sequences
1 µg of genomic DNA was diluted in ultra-pure water to 35 ng/μL and then sonicated in cold water with a Bioruptor sonicator (Diagenode) to obtain fragments averaging 300 bp in size. The fragmented DNA was used in combination with the hydroxymethyl collector (Active Motif) following the manufacturer’s protocol. Briefly, a glucose moiety that contains a reactive azide group was enzymatically linked to hydroxymethylcytosine in DNA, creating glucosyl-hydroxymethylcytosine. Next, a biotin conjugate was chemically attached to the modified glucose via a ‘click reaction’, and magnetic streptavidin beads were used to capture the biotinylated-hmC DNA fragments. After extensive washing steps and chemical elution, the hydroxymethylated DNA fragments released from the beads were used in sequencing experiments.
The library preparation was performed using the TruSeq ChIP Sample Prep Kit (Illumina). Briefly, double-stranded DNA was subjected to 5ʹ and 3ʹ protruding ends repair and non-templated adenines were added to the 3ʹ ends of the blunted DNA fragments to allow ligation of multiplex Illumina’s adapters. DNA fragments were then size selected (300–500bp) in order to remove all non-ligated adapters. 18 cycles of PCR were done to amplify the library which was then quantified by fluorometry using the Qubit 2.0 and its integrity was assessed with 2100 bioanalyzer (Agilent) before being sequenced. 6pM of DNA library, spiked with 1% PhiX viral DNA, were clustered on cBot (Illumina) and sequencing was performed on a HiScanSQ module (Illumina).
Validation of genomic data was performed by hMeDIP-qPCR using a specific anti-5hmC antibody and irrelevant IgG as a control. Input, hMeDIP and IgG products were used as templates for quantitative real-time PCR in a Roche LightCycler® system. The relative enrichment was calculated using the comparative CT method, which normalizes the amount of target to the input. Control regions monitored are as follows: ctrl 1 region is Dhodh gene (on chr8); ctrl 2 region is Olfr1178 gene (on chr2). Primers are available on request.
Library preparation, deep sequencing workflow and data analyses
The BWA software was used to map sequencing reads to the mouse genome (NCBI Build 37/UCSC mm9). Reads not uniquely mapped to the reference genome were discarded. Read density was computed by removing duplicate reads. To obtain sequencing tracks, bedGraph files (genomeCoverageBed) were uploaded onto the IGV genome browser [Citation76].
Bioinformatic analysis
To identify the differentially hydroxymethylated regions, the genome was first structured in fixed windows. The normalized 5hmC levels were then estimated by computing the fragment per kilobase million (FPKM) for every window and each condition. We eventually selected an optimal window-size of 5000bp (base pair) for our analysis according to the read density depth of the datasets and qPCR validation assays. The regions (windows of 5000bp) were then ranked based on their fold change and relative difference. Regions with an absolute fold change > 2 and absolute difference of at least 1 were selected for downstream analysis. Regions were related to genomic features using the VISTA Enhancer database (https://enhancer.lbl.gov), UCSC RefSeq and CpG islands annotations from genome reference mm9 and by computing the genomic overlap between the region centre and those features (an intersection of 1 base pair was considered positive). The genomic regions between the transcription start site (TSS) and the transcription termination site (TTS) as defined as Gene Body, the 2kb regions upstream the TSS was defined as the promoter. Regions ambiguously overlapping multiple features were associated with multiple categories. For the metagene analysis all the RefSeq genes were used to compute the relative average 5hmC signal inside the transcript (TSS to TTS).
Public databases have been used to retrieved histones marks (GSE90893) and gene expression (GSM1581307) data in MEFs and ESCs respectively. Transcriptomic analyses for MEFs Lsh KO are downloaded from E-MEXP-2383 and methylation array from E-MEXP-2385 [Citation77]. hMeDIP datasets produced from WT and Tet1 and Tet2 double knock (DKO) ESCs were retrieved from GSE72481 [Citation78]. TAB-seq data were downloaded from GSE1816853 [Citation79]. Sequenced files produced in this study on WT and Lsh KO MEFs and ESCs were uploaded on the GEO website under accession number GSE110129.
For clustering analysis, ChIP-sequencing datasets of histones marks were downloaded from GSE90893 and aligned onto the reference genome mm9. Occupancy of 7 histones modifications (H3K4me3, H3K4me2, H3K9Ac, H3K27Ac, H3K27me3, H3K79me2, H3K36me3) and one histone variant (H3.3) on the regions with differential hydroxymethylation were then visualized with the seqMINER software [Citation80]. Free clustering was done and the count in each category, defined by specific histone modification combination profile, was performed.
To evaluate the significance of the intersections between different datasets ‘Random’ distributions were generated using the ‘ShuffleBed’ option of BEDTools.
Repetitive element analysis (Pseudogenome)
A pseudogenome was generated with mouse DNA repeats sequences from RepeatMasker (http://www.repeatmasker.org/). 5hmC sequencing reads were mapped on this pseudogenome, using bowtie allowing two mismatches and without keeping reads mapped to more than one site. Duplicated reads were removed using samtools, and total reads mapped to each DNA repeats were calculated using samtools. The total numbers of reads mapped to each repeat element were normalized to the number of reads sequenced for each sample. To assess the effective change between the Lsh KO and the WT cells, the log odds ratio and P-value using a Fisher exact test were computed for each repetitive element. The P-values were then corrected using the FDR correction method [Citation81].
RT-qPCR and gene expression
Total RNA was extracted with the RNeasy Mini kit (Qiagen). After DNase I treatment (DNA-free DNase kit, Ambion), Superscript II reverse transcriptase (Invitrogen) were used to reverse-transcribe mRNAs to cDNAs. Gene expression levels were then evaluated by real-time PCR (LightCycler 480, Roche). Primers used to monitor Lsh, Tet1, Tet2 and Tet3 expression are available upon request.
Gene ontology annotation
Functional annotation of 5hmC-associated genes was performed using the MouseMine web interface [Citation82] using the Bonferroni Holm correction method, p-value<0.05 was considered significant. Gene ontology (Biological process) and Mammalian Phenotype ontology annotation were performed on December 2020 on lists of genes associated with gain or loss of 5hmC separately. A window with differential 5hmC was assigned to a gene when it overlapped with the region encompassing −2kb from the TSS up to the TTS.
LSH ChIP-sequencing analysis
LSH ChIP-sequencing data in MEFs were retrieved from GEO database with accession number GSM835828 [Citation74]. LSH binding sites were intersected with the list of differentially 5hmC regions to identify overlapping (at least 1bp) and closest LSH sites from a differentially 5hmC region.
Results
LSH is a TET-interacting factor
To explore the mechanisms of action of TET proteins, we used the already described HaloTag technology followed by mass-spectrometry analysis, in order to identify proteins co-immunoprecipitating with Halo-tagged-TET1, -TET2 and -TET3 protein expressed in human HEK293T cells [Citation29]. We identified 56 candidate interaction partners alongside the O-Linked N-Acetylglucosamine Transferase (OGT) [Citation29], the Poly(ADP-Ribose) Polymerase 1 (PARP1) [Citation22] and the Proliferating Cell Nuclear Antigen (PCNA) [Citation83] proteins, already described as TET proteins partners. The chromatin-remodelling protein LSH was identified as one of the 56 candidates interacting with TET1 and TET2 and TET3.
We further explored the interaction between TET proteins and LSH by semi-endogenous and endogenous co-IPs. HEK293T cells were transfected with Flag-tagged human TET1, TET2 or TET3 catalytic domain (CD) and the empty vector. We found by co-immunoprecipitation that endogenous LSH interacts with Flag-tagged CDs of TET1, TET2 and TET3 but not with the Flag control (). For the reverse approach, we immunoprecipitated LSH and confirmed by western blot the presence of Flag-tagged CDs of TET proteins in LSH co-immunoprecipitates (Supplemental ).
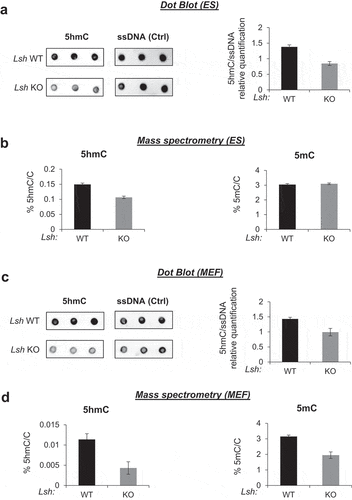
Figure 2. Lsh knock-out decreases 5hmC global level in ES and MEF cells.
To map the domains of LSH interacting with TET proteins, we performed in vitro GST pull-down assays. We used full-length LSH produced in bacteria as well as truncated forms of the protein, including the LSH coiled-coil (or CC; amino-acids 1–226), LSH DEXH-box helicase domain containing the ATPase domain (aa 227–589) and LSH C-terminal domain (or CT; aa 590–838) [Citation74]. TET full-length and CD domain were produced by in vitro transcription/translation. We observed that TET proteins and CD domains interact in vitro with the coiled-coil domain (CC) of LSH (). We conclude that a sequence encompassing the CD of TETs is sufficient to bind the CC domain of LSH in vitro.
We then investigated the interaction between endogenous TETs and LSH in mouse ESCs using antibodies specific for LSH (), for TET1 (left panel) and TET2 (right panel) (uncropped western blots are available in Supplemental Supplemental ). We did not assess for TET3/LSH interaction in ESCs because TET3 is expressed at low levels in this cell type [Citation84,Citation85]. Our results indicated endogenous interactions in ESCs between LSH and TET1, and between LSH and TET2. Our data indicate that LSH and TET proteins interact in vitro and in vivo.
Subsequently, we wondered whether in addition to interacting with TET enzymes, LSH was also regulating their mRNA expression in ESCs and MEFs. We thus conducted RT-qPCR in Lsh KO ESCs and MEFs, as well as in KD ESCs. We observed that Tet1 and Tet2 mRNA expression is lower in Lsh KO and KD ESCs compared to their respective control ESCs (Supplemental ). In MEFs Tet1 and Tet2 mRNA expression is also lower, with a statistically significant p-value in Lsh KO compared to control (Supplemental ). No statistically significant differences were observed for Tet3 in both ESCs and MEFs. Taken together, these data suggest that LSH might regulates 5hmC amount through different mechanisms in ESCs and MEFs (including the binding to TET enzymes and/or the regulation of their mRNA expression) that may impact both global and local 5hmC levels.
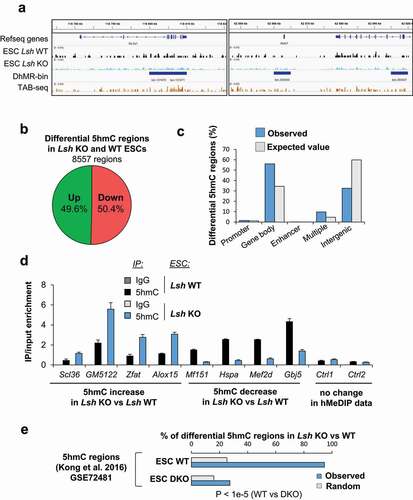
Figure 3. 5hmC level is impaired in Lsh KO ESCs at specific genomic loci.
LSH knock-out impairs the global hydroxymethylation levels in MEFs and ESCs
We tested whether LSH was essential for 5hmC in ESCs and MEFs. To explore the role of LSH in 5hmC, we performed dot blot experiments and mass spectrometry (MS) analyses (). We prepared genomic DNA samples from Lsh KO and WT ESCs and analysed them by dot blot with anti-5hmC and anti-single stranded DNA antibodies as a control. The relative-quantification of 5hmC signals relative to total DNA using the ImageJ software showed that the levels of 5hmC are lower in Lsh KO compared to WT ESCs (). We also analysed the samples by MS and confirmed the lower levels (29%) of 5hmC in the absence of Lsh (). In addition, MS analysis revealed that levels of 5mC were similar in Lsh KO and WT ESCs (). We validated these observations by showing that depletion of Lsh by short-hairpin RNA (shRNA) in ESCs also causes 50% (KD1), 32% (KD2) and 70% (KD3) reduction in 5hmC levels in the DNA compared to control (Supplemental ). Interestingly, in ESCs we also observed a 15-fold reduction of another oxidized form of 5mC, the 5-formylcytosine (5fC) (Supplemental ). In sum, we observed that LSH is required to maintain 5hmC levels in the DNA in ESCs.
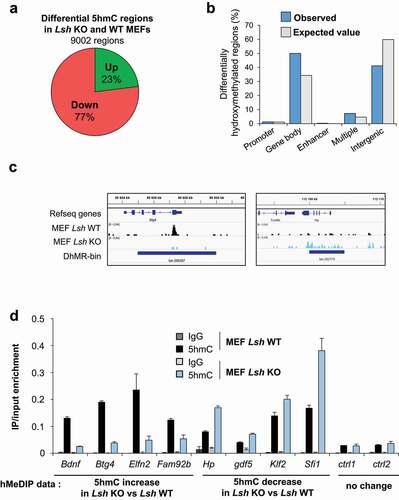
Figure 4. 5hmC level is impaired in Lsh KO MEFs cells at specific genomic loci.
We then investigated the role of LSH in MEFs. Our MS data confirmed that the levels of 5hmC are 10 times higher in ESCs than in MEFs or differentiated cells, as already observed [Citation86]. In MEFs, we observed by dot blot that 5hmC levels were 1/3 lower in Lsh KO MEFs compared to WT MEFs (). Again, MS confirmed this result by showing a 63% reduction of 5hmC (). As previously described, we also observed a reduction (by 40%) in the levels of 5mC by MS in the absence of Lsh () [Citation58,Citation77,Citation87].
Altogether, our results suggest that LSH maintains and/or establishes 5hmC levels in ESCs and MEFs. In ESCs, we could not detect significant changes in 5mC amount in the absence of Lsh suggesting that LSH could directly affect the function of TET enzymes and 5hmC patterns. On the contrary, in MEFs, LSH regulates both 5mC and 5hmC levels suggesting that the role of LSH in 5hmC regulation might be partially related to its known function in 5mC establishment and/or maintenance.
5hmC changes at the genome-wide level in Lsh KO ESCs, mostly in gene bodies
To further explore whether LSH regulates 5hmC levels at specific loci in the genome, we mapped 5hmC in Lsh KO and WT ESCs and MEFs. To map 5hmC in the genome we used a simple and efficient glucosylation reaction procedure followed by deep sequencing. We then used a window-based approach (5,000 base pairs) to analyse the read density along chromosomes and we compare the reads density in Lsh KO and WT cells.
We identified 8557 windows showing differential 5hmC levels in Lsh KO ESCs compared to WT ESCs, as shown for the representative gene Slc2a1 and Atoh7 (). These regions, as exemplified by TAB-seq data, contain numerous 5hmCpG sites (, Supplemental ) [Citation79]. 4312 regions exhibit reduced levels of 5hmC and 4245 regions show increased levels of 5hmC in Lsh KO ESCs, corresponding to 5017 genes. We found 2659 genes with gain of 5hmC and 2448 genes with reduced levels of 5hmC (Supplemental Table 1). A deeper analysis of the genomic distribution of these differentially 5hmC regions show enrichment at promoters (124/96 expected), gene bodies (4801/2940 expected), enhancers (13/5), multiple regions (i.e. region with at least two different genomic features, e.g. promoter and enhancer) (830/395 expected) and an under-representation in intergenic regions (2789/5122 expected) () (p < 0.00001; χ2 goodness of fit). These data indicate that changes in 5hmC levels in Lsh KO occur locally in gene bodies, promoters and enhancers more than expected by chance ( and Supplemental ).
Figure 5. 5hmC differences are not a direct consequence of 5mC modifications upon Lsh loss and are not always followed by direct gene expression change.
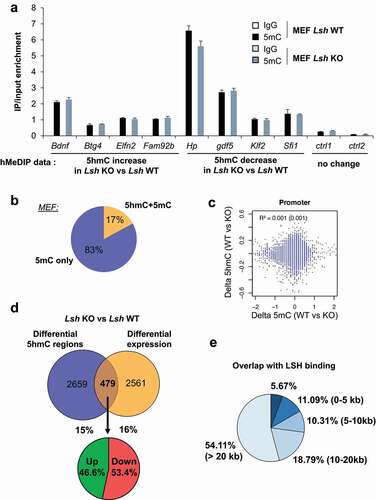
A functional analysis of genes associated with hyper- and hypo-hydroxymethylated regions revealed significant over-representation of pathways associated with development, such as ‘tissue development’, ‘embryonic development’ and ‘organismal development’ (Supplemental ). We validated the changes in 5hmC profile at some of these genes of interest by an orthogonal approach. Using a DNA pull-down approach with a specific 5hmC-antibody, or an irrelevant IgG as a control, we confirmed that 5hmC level was increased at Scl36, GM5122, Zfat and Alox15 genes, while it was reduced at Mf151, Hspa, Mef2d and Gbj5 genes (), as observed in the genomic analysis with the glucosylation reaction procedure. No changes of 5hmC were detected at two different control regions using this hMeDIP-qPCR analysis ().
We then analysed the profile of 5hmC levels on the genes showing reduced and increased levels of 5hmC in Lsh KO with a metagene analysis (Supplemental ). We observed that in control cells, on average, the levels of 5hmC were higher in the group of genes with differential 5hmC levels upon Lsh KO compared to the entire set of mouse genes (Supplemental and ). Nonetheless, no differences were observed between the group of genes loosing and gaining 5hmC in Lsh KO (Supplemental ). A focus on transcription start sites (TSS) and transcription termination sites (TTS) showed a slight accumulation of 5hmC 1kb upstream to the TSS for genes gaining 5hmC upon Lsh KO (Supplemental ).
We then investigated whether these changes in 5hmC fall in the same regions of those with loss of 5hmC upon Tet1 and Tet2 double knock (DKO) in ESCs [Citation78]. We intersected hMeDIP mapping analyses in WT and Tet1/2 DKO ESCs with our list of 8557 DhMRs. We observed that virtually all DhMRs (94.6%) lies in 5hmC domains defined by hMeDIP-sequencing in WT ESCs (respective control of DKO ESCs). On the contrary, Tet1/2 DKO hydroxymethylome in ESCs overlap only at 27% with the DhMRs (). This result suggests that most of the 5hmC regions regulated by LSH identified in our analysis overlap with 5hmC domains regulated by TET1 and TET2 enzymes in ESCs. This is consistent with the existence of a LSH/TET axis in 5hmC regulation.
5hmC changes at the genome-wide level in Lsh KO MEFs
We then mapped and analysed the distribution of 5hmC in Lsh KO and WT MEFs. We identified 9002 regions, corresponding to 3138 genes, with differential hydroxymethylation in Lsh KO MEFs compared to control MEFs. 77% of the regions (n = 6932) showed loss of 5hmC and 23% of the regions (n = 2070) gain of 5hmC () in Lsh KO MEFs, corresponding to 2376 and 1043 genes, respectively (Supplemental table 1). These changes in 5hmC levels occur predominantly in gene bodies and intergenic regions and are quite rarely found at promoters and/or CpG islands, as illustrated for Btg4 and Hp genes ( and Supplemental Figure 6A). Nonetheless, these observed changes in promoters, gene bodies and enhancers occur more frequently than expected by chance (p < 0.00001; χ2 goodness of fit) ().
Changes in 5hmC level detected by high throughput sequencing were further investigated by hMeDIP-qPCR. This analysis confirmed that Bdnf, Btg4, Elfn2 and Fam92 harbour an increase of 5hmC whereas Hp, Gdf5, Klf2 and Sfi1 showed a decrease of 5hmC in Lsh KO MEFs compared to WT MEFs, as observed in the genomic analysis (). No change of 5hmC was detected at control regions (). We then investigated the ‘metagene’ profile of 5hmC in MEFs, and observed again that genes showing changes in 5hmC present higher levels of 5hmC than averaged mice genes (Supplemental Figure 6 C). No significant pattern is observed between genes gaining and/or loosing 5hmC at their TSS, gene body and TTS in control cells (Supplemental Figure 6D). Functional analysis of differentially hydroxymethylated genes showed enrichment in GO term ‘preweaning lethality’, ‘nervous system development’ and ‘abnormal homeostasis’ (Supplemental Figure 6 C) highlighting again the link between LSH, 5hmC and developmental processes [Citation55,Citation88,Citation89]. Importantly, GO term enrichments were quite similar for genes with gain or loss of 5hmC (Supplemental Figure 6D).
Lsh regulates 5hmC at repetitive sequences
A common finding in the literature is the role of LSH at repeated minor satellite sequences [Citation68,Citation87,Citation90,Citation91]. We thus directly accessed the levels of 5hmC at major and minor satellite as well as repeated sequences such as LINE1 and SINE1 (Supplemental Figure 7A). We observed by hMeDIP-qPCR a decrease in 5hmC levels at minor and major satellites as well as LINE1 elements in Lsh KO MEFs compared to WT MEFs. We also detected a weak increase in 5hmC levels at SINE elements in Lsh KO MEFs compared to control (Supplemental Figure7A).
We further expanded the analysis to all DNA repeated sequences by mapping 5hmC-enriched sequencing reads on a synthetic pseudogenome containing all the repeated elements of the mice genome (Supplemental Figure 7B). We identified several classes and families of repeated DNA elements that exhibit changes in 5hmC levels between Lsh KO MEFs and ESCs compared to controls. We observed increased levels in 5hmC at L1 elements and decreased levels of 5hmC rRNAs in Lsh KO MEFs (Supplemental Figure 7B). In ESCs, we observed reduced levels of 5hmC at major and minor satellites, as well as L1 and Long Terminal Repeats (LTRs) sequences, in Lsh KO ESCs compared to control ESCs (Supplemental Figure 7B).
Thus, besides gene-bodies, LSH regulates 5hmC levels at specific repetitive DNA sequences in ESCs and MEFs.
Relationship between LSH binding, DNA modifications and gene expression in MEF cells
Previous studies have characterized the consequences of Lsh KO on the landscape of 5mC and gene expression in the same MEF cell lines we utilized for our analysis [Citation77] as well as the distribution of LSH-binding sites [Citation74]. Using this information, we addressed the relationship between changes in 5hmC at specific genes, LSH binding and their relation to gene expression.
We first wondered whether differentially 5hmC regions were associated with changes in 5mC. We conducted hMeDIP- and MeDIP-qPCR analyses at specific regions identified in our analysis. We observed that 5hmC changes observed at specific sites, were not associated with changes in 5mC ( and ). To further explore this point we re-analysed the map of 5mC at the promoter of mouse genes in Lsh KO and control MEFs [Citation77]. We found that only 17% of promoters with differential 5hmC overlap with promoters with 5mC changes upon Lsh KO. This overlap was not statistically significant and overall there is no correlation between changes in levels of 5hmC and 5mC at promoters in Lsh KO MEFs (). These data indicate that the patterns of differentially methylated regions and hydroxymethylated regions at promoters are different. These findings suggest that the impact of LSH in the regulation of 5hmC patterns is not directly correlated to its function on 5mC in MEFs.
Myant and colleagues performed a global analysis of the transcriptionally mis-regulated genes in Lsh KO versus WT MEFs cells using a microarray platform [Citation77]. We used these data to compare the regions showing differential 5hmC with expression data (Supplemental table 1). We found that only 15% of genes that exhibit differential 5hmC levels also have an alteration of gene expression and 16% of genes that harbour differential expression have alteration of 5hmC level and the overlap between the two was not significant (). Also, no global expression changes were found between gene with deregulated 5hmC to all others (same results were obtained with the analysis on hypo- and hyper-hydroxymethylated genes separately) (Supplemental Figure 8A). Thus, we confirmed that at the genome-wide level, changes in 5hmC landscape around genes mildly correlate with changes in gene expression. This observation is reminiscent of previous studies showing a poor overlap (~10%) between loss or gain of DNA methylation and gene expression changes in Lsh KO compared to WT MEFs [Citation77]. A gene ontology analysis, on these 479 genes showing both changes in 5hmC and expression levels in Lsh KO MEFs, reveals an enrichment for cellular functions associated with ‘cell movement’, ‘cell morphology’, ‘cell survival and death’ (Supplemental Figure 8B).
Subsequently, we compared the 9002 DhMR regions to histones marks and H3.3 variant that are characteristics of different chromatin environment and genomic features such as promoters (H3K4me3 and H3K9Ac), active promoters and enhancers (H3K4me2 and H3K27Ac), transcription (H3K36me3 and H3K79me2) and repressive compartments (H3K27me3) [Citation92] (Supplemental Figure 6B). 21% of the 9002 regions correlated with active histones marks: H3K27Ac, H3K4me2/3, H3K9Ac (group 1 and 2), 19% correlated with elongating marks H3K36me3 and/or H3K79me2 (group 3 and 4) and only 10% with repressive marks, H3K27me3 (group 5). Intriguingly, we found that half of the regions identified in our study do not harbour any examined marks (group 6). These data confirm that changes in 5hmC upon Lsh KO predominantly occur in intergenic regions and gene bodies. The data also suggest that there is no clear relationship between histone modifications in MEFs and changes in 5hmC levels in Lsh KO MEFs. Nonetheless, most analysed regions fall in transcribed domains that present high levels of marks associated with gene bodies, such as H3K36me3.
We finally explored whether the changes in 5hmC were associated with the binding of LSH. We re-analysed a ChIP-sequencing analysis of LSH in MEFs [Citation74]. We observed that LSH binds at a number of differentially 5hmC regions identified in Lsh KO MEFs (). Nonetheless, in most cases, no LSH binding is observed in the vicinity of the differentially 5hmC region, suggesting transient or dynamic binding of LSH in these regions ().
Discussion
LSH interacts with TET enzymes
The chromatin remodelling protein LSH is involved in chromatin organization, gene expression and DNA methylation. Several studies have shown that LSH interacts with DNMTs [Citation56,Citation61] and that LSH regulates DNA methylation levels, in particular at DNA repetitive sequences in MEFs [Citation57,Citation62]. Here we show that LSH can also interact with TET proteins to establish and/or maintain the levels of 5hmC in the DNA, globally but also at specific genomic sites.
We show that LSH co-immunoprecipitates with TET proteins in human and murine cells, and that it binds the catalytic domain of TET proteins in vivo and in vitro. We also document that TET proteins interact with the coiled-coil (CC) domain of LSH. CC domains are often involved in protein–protein interaction and this region of LSH is required for transcriptional silencing independently of its chromatin-remodelling activity [Citation61]. Surprisingly, DNMTs interact with the CC domain of LSH [Citation61], indicating that TETs and DNMTs both interact with a similar region of LSH. This observation raises intriguing questions regarding the regulation of DNA methylation/demethylation processes by LSH. Additional experiments are needed to clarify whether the binding of TETs and DNMTs to LSH are mutually exclusive or whether post-translational modifications regulate the kinetic of TETs and DNMTs binding during cell fate transitions.
Importantly, the CC domain of LSH also interacts with additional proteins, such as the Pyruvate Kinase M2 (PKM2), the transcription factor E2F3 and histone deacetylases (HDACs). The LSH/PKM2 complex regulates the transactivation of transcription factor p53 [Citation93]. The binding of LSH to E2F3 regulates its transcriptional activity in cancer cells [Citation74], and it may be important for the maintenance of cancer stem cells [Citation94]. The LSH/HDACs interaction contributes to gene silencing, independently of LSH chromatin remodelling activity [Citation61]. Therefore, it will thus be necessary to understand whether different complexes involving LSH co-exist in cells and at genomic targets to regulate chromatin organization and gene expression.
Our analysis also unveils that LSH regulates the expression of TET enzymes in ESCs and MEFs. Our data indicate that LSH might regulate 5mC and 5hmC amount and distribution in the genome at multiple levels in murine cells, by regulating the amount of TET enzymes and possibly their recruitment at specific sites in the genome. Further work will be needed to understand the contribution of each axis in 5hmC establishment and/or maintenance.
LSH participates in the regulation of the amount and distribution of 5hmC
Many studies have investigated the role of chromatin-associated factors and transcription factors in the establishment and removal of 5hmC, as well as the positioning of this epigenetic mark [Citation10,Citation95–98]. One of these studies reported that LSH binds DNA oligo-nucleotides containing 5hmC in vitro, while it has a weaker affinity for the same oligonucleotide containing 5mC or C instead of 5hmC [Citation10]. Another study showed that, in a human renal cancer cell model, LSH induces the expression of TET enzymes and affects 5hmC levels during the course of cancer progression [Citation48]. In our study, we showed that LSH and TET proteins directly interact and that changes in 5hmC levels occur in defined regions of the genome. Taken together, these independent studies suggest that LSH is involved in 5hmC signalling and that it likely modulates 5hmC levels in different cell types through different mechanisms.
In ESCs and MEFs we demonstrated that LSH regulates the global pattern of 5hmC by dot-blot, MS and deep sequencing experiments. Loss of LSH causes a dramatic reduction in levels of 5hmC in the DNA, and at least in ESCs, this reduction is not mimicked by a loss of 5mC. This observation suggests a direct role of LSH in the establishment and/or maintenance of 5hmC pattern in ESCs, which has not been previously proposed. Furthermore, at the genomic scale, promoters with 5mC changes do not correspond to changes in 5hmC in Lsh KO MEFs, further supporting a potential direct role of LSH in 5hmC biology.
Our data clearly suggest that LSH is a central regulator of DNA modifications by influencing not only DNA methylation but also, to a lesser extent, DNA hydroxymethylation. Intriguingly, since TET enzymes interact with the CC domain of LSH it is still unclear whether this new function of LSH in 5hmC signalling involved its chromatin-remodelling activity. We observed that most regions with differential 5hmC upon Lsh KO are located away from a strong LSH binding site detectable by ChIP-sequencing. In cases of transient or dynamic binding of the enzyme onto the DNA, it is not uncommon to detect a mark but not the enzyme depositing this mark (or a co-regulator) [Citation99]. Consistent with this possible interpretation, TET enzymes and 5hmC domains do not perfectly overlap in ESCs [Citation100]. The lack of correlation between LSH binding sites and DhMRs could also indicate that the interaction between LSH and TET enzymes affect other functions of TETs, unrelated to 5hmC. TET enzymes exhibit several non-catalytic functions in gene expression and in the regulation of pluripotency in the haematologic lineage [Citation101,Citation102]. The LSH/TET axis might thus have additional functions not investigated in this study. For instance, TETs and LSH may play a role on the regulation of histone mark deposition and maintenance as well as DNA repair mechanisms [Citation67,Citation103–106].
LSH in gene expression, ESC pluripotency and disease
While 5mC and 5hmC function is well understood at promoter of genes, it still remains unclear how changes in 5hmC in Lsh KO cells relate to gene expression regulation. ESCs transition through different pluripotency states and it is known that TET enzymes might contribute to the regulation of this process. For instance, TET1 is expressed in both naive and primed ESCs while TET2 is only expressed in naive ESCs [Citation107]. We observed that changes in 5hmC levels upon Lsh KO are not sufficient to disorganize the transcriptional network of ESCs, and only moderately affect overall gene expression. Consistently, the disruption of Lsh in ESCs had no apparent effect on the maintenance of pluripotency (our observations and [Citation58,Citation91]). However, changes in 5hmC could generate a more permissive state that could facilitate future transcriptional induction. It would be interesting to further investigate the dynamics of 5hmC and gene expression when Lsh KO ESCs differentiate into different lineages (ectoderm, mesoderm and endoderm). Consistently with this hypothesis, we observed that the genes affected by Lsh KO are enriched for ‘developmental’ and ‘cell differentiation’ genes. Hence, changes in 5hmC at specific genes in ESCs might have a moderate impact on gene expression when ESCs are maintained in LIF+serum condition (this study), but would be mis-regulated if ESCs are induced to differentiate.
The genes regulated by the LSH/5hmC signalling identified in our analysis may also provide a valuable insight in the understanding of LSH function in diseases. LSH is located in a break point region frequently associated with leukaemia [Citation50] and a deletion in LSH gene is found in 57% of acute myeloid leukaemia (AML) and 37% of acute lymphocytic leukaemia (ALL) patients [Citation108]. Interestingly, 5hmC is often deregulated in haematological malignancies and TET2 is one of the most mutated genes in leukaemia. In mice, loss of Tet2 increases the haematopoietic stem cell compartment and skews cell differentiation towards the myeloid compartment [Citation109,Citation110]. Both 5hmC levels and LSH protein levels are also reduced in several solid tumours, such as nasopharyngeal carcinoma, breast or colon cancer [Citation48]. LSH, as well as DNMT genes, is also mutated in immunodeficiency, centromeric region instability and facial abnormalities (ICF) syndrome [Citation53,Citation111–113]. The link between LSH, TETs and 5hmC is not yet fully explored in ICF and cancers and our lists of differentially hydroxymethylated genes could be a valuable tool to design new diagnostic and therapeutic strategies.
In summary, we report an interaction between TET proteins and LSH, and provide evidence that LSH is a regulator of DNA hydroxymethylation. This information clearly contributes to a better understanding of the crosstalk between chromatin organization, DNA modifications and gene expression.
Accession numbers
hMe enriched DNA-sequencing on WT and Lsh KO MEFs and ESCs were uploaded on GEO servers under the GSE110129 number.
Authors contributions
MdD, RD and FF designed and coordinated the study. MdD, LC, EC run the experiments. MBi performed the bioinformatics analyses. MBa performed the MS experiments. CL and IS designed and generated the Lsh KO ESCs. MdD, LC, RD and FF wrote the manuscript. MdD, MBa, IS, BM and FF edited and revised the manuscript. All the authors edited and approved the final manuscript and Dr Matthieu Gérard (Université Paris-Saclay, Gif-sur-Yvette, France) for technical help with the ESCs culture in his lab.
Supplemental Material
Download Zip (2.6 MB)Acknowledgements
We thank Dr Kathrin Muegge (Center for Cancer Research; National Cancer Institute; Frederick, MD; USA) for the generous gift of MEFs Lsh WT and KO. We thank Dr Ulrike Ziebold (Cancer Research, Berlin, Germany) for the generous gift of Lsh plasmids. We thank Dr Matthieu Defrance (Université Libre de Bruxelles, Belgium) and Dr Gwenneg Kerdivel (Cochin Institute, Paris, France) for bio-informatical analyses as well as Dr Claire Marchal (National Institute of Health, Bethesda, USA) for the technical help for repetitive region analysis. We thank Dr Caterina Lucano for critical reading of the manuscriptand Dr Matthieu Gérard (Université Paris-Saclay, Gif-sur-Yvette, France) for technical help with the ESCs culture in his lab.
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20(24):3139–3155.
- Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011;12:647–656. Nature Publishing Group
- Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends Biochem Sci. 2011;36(7):381–387.
- Loaeza-Loaeza J, Beltran AS, Hernández-Sotelo D. DNMTs and impact of CpG content, transcription factors, consensus motifs, lncRNAs, and histone marks on DNA methylation. Genes (Basel). 2020;11(11):11.
- Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935.
- Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES cell self-renewal, and ICM specification. Nature. 2010;466(7310):1129–1133.
- Kriaucionis S, Heintz N. The nuclear DNA base, 5-hydroxymethylcytosine is present in brain and enriched in Purkinje neurons. Science. 2009;324(5929):929–930.
- Schomacher L, Niehrs C. DNA repair and erasure of 5-methylcytosine in vertebrates. BioEssays News Rev Mol Cell Dev Biol. 2017;39(3).
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479.
- Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–1159.
- Bachman M, Uribe-Lewis S, Yang X, et al. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem. 2014;6(12):1049–1055.
- Etchegaray J-P, Chavez L, Huang Y, et al. The histone deacetylase sirt6 controls embryonic stem cell fate via Tet-mediated production of 5-Hydroxymethylcytosine. Nat Cell Biol. 2015;17(5):545–557.
- Uribe-Lewis S, Carroll T, Menon S, et al. 5-hydroxymethylcytosine and gene activity in mouse intestinal differentiation. Sci Rep. 2020;10(1):546.
- Ma L, Muhammad T, Wang H, et al. Putative promoters within gene bodies control exon expression via TET1-mediated H3K36 methylation. J Cell Physiol. 2020;235(10):6711–6724.
- Tekpli X, Urbanucci A, Hashim A, et al. Changes of 5-hydroxymethylcytosine distribution during myeloid and lymphoid differentiation of CD34+ cells. Epigenetics Chromatin. 2016;9(1):21.
- Tsagaratou A, Äijö T, Lio C-WJ, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A. 2014;111(32):E3306–3315.
- Pastor WA, Pape UJ, Huang Y, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473(7347):394–397.
- Song C-X, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29(1):68–72.
- Rausch C, Hastert FD, Cardoso MC. DNA modification readers and writers and their interplay. J Mol Biol. 2019. DOI:https://doi.org/10.1016/j.jmb.2019.12.018
- Ko M, An J, Bandukwala HS, et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497(7447):122–126.
- Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, et al. Nanog-dependent function of Tet1 and Tet2 in establishment of pluripotency. Nature. 2013;495(7441):370–374.
- Ciccarone F, Valentini E, Zampieri M, et al. 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget. 2015;6(27):24333–24347.
- De La Rica L, Rodríguez-Ubreva J, García M, et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013;14(9):R99.
- Guilhamon P, Eskandarpour M, Halai D, et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. 2013;4(1):2166.
- Okashita N, Kumaki Y, Ebi K, et al. PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Dev Camb Engl. 2014;141:269–280.
- Kienhöfer S, Musheev MU, Stapf U, et al. GADD45a physically and functionally interacts with TET1. Differ Res Biol Divers. 2015;90(1–3):59–68.
- Collignon E, Canale A, Al Wardi C, et al. Immunity drives TET1 regulation in cancer through NF-κB. Sci Adv. 2018;4(6): eaap7309.
- Cheng Z-L, Zhang M-L, Lin H-P, et al. The Zscan4-Tet2 transcription nexus regulates metabolic rewiring and enhances proteostasis to promote reprogramming. Cell Rep. 2020;32(2):107877.
- Deplus R, Delatte B, Schwinn MKMK, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. Embo J. 2013;32(5):645–655.
- Cartron P-F, Nadaradjane A, Lepape F, et al. Identification of TET1 partners that control its DNA-demethylating function. Genes Cancer. 2013;4(5–6):235–241.
- Song C, Wang L, Wu X, et al. PML recruits TET2 to regulate DNA modification and cell proliferation in response to chemotherapeutic agent. Cancer Res. 2018;78(10):2475–2489.
- Cheng J, Guo S, Chen S, et al. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5(2):471–481.
- Fu X, Jin L, Wang X, et al. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci U S A. 2013;110(44):17892–17897.
- Morita S, Horii T, Kimura M, et al. miR-29 represses the activities of DNA methyltransferases and DNA demethylases. Int J Mol Sci. 2013;14(7):14647–14658.
- Song SJ, Poliseno L, Song MS, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154(2):311–324.
- Lv X, Jiang H, Liu Y, et al. Micro RNA-15b promotes neurogenesis and inhibits neural progenitor proliferation by directly repressing TET 3 during early neocortical development. EMBO Rep. 2014;15(12):1305–1314.
- Rao VK, Swarnaseetha A, Tham G-H, et al. Phosphorylation of Tet3 by cdk5 is critical for robust activation of BRN2 during neuronal differentiation. Nucleic Acids Res. 2020;48(3):1225–1238.
- Zhang T, Guan X, Choi UL, et al. Phosphorylation of TET2 by AMPK is indispensable in myogenic differentiation. Epigenetics Chromatin. 2019;12(1):32.
- Wu D, Hu D, Chen H, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018;559(7715):637–641.
- Ito R, Katsura S, Shimada H, et al. TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells Devoted Mol Cell Mech. 2014;19(1):52–65.
- Zhang Q, Liu X, Gao W, et al. Differential regulation of the Ten-Eleven Translocation (TET) Family of Dioxygenases by O-Linked β-N-Acetylglucosamine Transferase (OGT). J Biol Chem. 2014;289(9):5986–5996.
- Bauer C, Göbel K, Nagaraj N, et al. Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-Linked N-Acetylglucosamine Transferase (OGT). J Biol Chem. 2015;290(8):4801–4812.
- Blaschke K, Ebata KT, Karimi MM, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500(7461):222–226.
- Chen J, Guo L, Zhang L, et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet. 2013;45(12):1504–1509.
- Minor EA, Court BL, Young JI, et al. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288(19):13669–13674.
- Yin R, Mao S-Q, Zhao B, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135(28):10396–10403.
- Hore TA, Meyenn F, Von, Ravichandran M, et al. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc Natl Acad Sci. 2016;113(43):12202–12207. National Academy of Sciences
- Jia J, Shi Y, Chen L, et al. Decrease in lymphoid specific helicase and 5-hydroxymethylcytosine is associated with metastasis and genome instability. Theranostics. 2017;7(16):3920–3932.
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78(1):273–304.
- Geiman TM, Durum SK, Muegge K. Characterization of gene expression, genomic structure, and chromosomal localization of Hells (Lsh). Genomics. 1998;54(3):477–483.
- Geiman TM, Muegge K. Lsh, an SNF2/helicase family member, is required for proliferation of mature T lymphocytes. Proc Natl Acad Sci U S A. 2000;97(9):4772–4777.
- Brzeski J, Jerzmanowski A. Deficient in DNA methylation 1 (DDM1) defines a novel family of chromatin-remodeling factors. J Biol Chem. 2003;278(2):823–828.
- Jenness C, Giunta S, Müller MM, et al. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc Natl Acad Sci U S A. 2018;115(5):E876–85.
- Dennis K, Fan T, Geiman T, et al. Lsh, a member of the SNF2 family, is required for genome-wide methylation. Genes Dev. 2001;15(22):2940–2944.
- Ren J, Briones V, Barbour S, et al. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015;43(3):1444–1455.
- Zhu H, Geiman TM, Xi S, et al. Lsh is involved in de novo methylation of DNA. Embo J. 2006;25(2):335–345.
- Huang J, Fan T, Yan Q, et al. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 2004;32(17):5019–5028.
- Xi S, Geiman TM, Briones V, et al. Lsh participates in DNA methylation and silencing of stem cell genes. Stem Cells Dayt Ohio. 2009;27(11):2691–2702.
- Tao Y, Xi S, Shan J, et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc Natl Acad Sci U S A. 2011;108(14):5626–5631.
- Ren J, Finney R, Ni K, et al. The chromatin remodeling protein Lsh alters nucleosome occupancy at putative enhancers and modulates binding of lineage specific transcription factors. Epigenetics. 2019;14(3):277–293.
- Myant K, Stancheva I. LSH cooperates with DNA methyltransferases to repress transcription. Mol Cell Biol. 2008;28(1):215–226.
- Dunican DS, Cruickshanks HA, Suzuki M, et al. Lsh regulates LTR retrotransposon repression independently of Dnmt3b function. Genome Biol. 2013;14(12):R146.
- Termanis A, Torrea N, Culley J, et al. The SNF2 family ATPase LSH promotes cell-autonomous de novo DNA methylation in somatic cells. Nucleic Acids Res. 2016;44(16):7592–7604. Oxford Academic
- Yu W, McIntosh C, Lister R, et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24(10):gr.172015.114.
- Yu W, Briones V, Lister R, et al. CG hypomethylation in Lsh−/− mouse embryonic fibroblasts is associated with de novo H3K4me1 formation and altered cellular plasticity. Proc Natl Acad Sci U S A. 2014;111(16):5890–5895.
- Yan Q, Huang J, Fan T, et al. Lsh, a modulator of CpG methylation, is crucial for normal histone methylation. Embo J. 2003;22(19):5154–5162.
- Ni K, Ren J, Xu X, et al. LSH mediates gene repression through macroH2A deposition. Nat Commun. 2020;11(1):5647.
- Sun L-Q, Lee DW, Zhang Q, et al. Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev. 2004;18(9):1035–1046.
- Geiman TM, Tessarollo L, Anver MR, et al. Lsh, a SNF2 family member, is required for normal murine development. Biochim Biophys Acta. 2001;1526(2):211–220.
- Berlivet S, Guiraud V, Houlard M, et al. pHYPER, a shRNA vector for high-efficiency RNA interference in embryonic stem cells. BioTechniques. 2007;42(738):740–743.
- Depaux A, Dieuleveult M, De, Hmitou I, et al. Tandem affinity purification of remodeler-mononucleosome complexes for ChIP-seq in mouse embryonic stem cells. 2014 [ cited 2019 Jan 4]; Available from: https://www.nature.com/protocolexchange/protocols/3459
- Schnütgen F, De-Zolt S, Van Sloun P, et al. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc Natl Acad Sci U S A. 2005;102(20):7221–7226.
- Deplus R, Brenner C, Burgers WA, et al. Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res. 2002;30(17):3831–3838.
- Von Eyss B, Maaskola J, Memczak S, et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. Embo J. 2012;31(4):972–985.
- Fuks F, Burgers WA, Brehm A, et al. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24(1):88–91.
- Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26.
- Myant K, Termanis A, Sundaram AYM, et al. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21(1):83–94.
- Kong L, Tan L, Lv R, et al. A primary role of TET proteins in establishment and maintenance of De Novo bivalency at CpG islands. Nucleic Acids Res. 2016;44(18):8682–8692.
- Von Meyenn F, Iurlaro M, Habibi E, et al. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol Cell. 2016;62(6):848–861.
- Ye T, Krebs AR, Choukrallah M-A, et al. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39(6):e35–e35.
- Deplus R, Blanchon L, Rajavelu A, et al. Regulation of DNA methylation patterns by CK2-mediated phosphorylation of Dnmt3a. Cell Rep. 2014;8(3):743–753.
- Motenko H, Neuhauser SB, O’Keefe M, et al. MouseMine: a new data warehouse for MGI. Mamm Genome. 2015;26(7–8):325–330. Off J Int Mamm Genome Soc
- Zheng L, Zhai Y, Li N, et al. The modification of Tet1 in male germline stem cells and interact with PCNA, HDAC1 to promote their self-renewal and proliferation. Sci Rep. 2016;6(1):37414.
- Dawlaty MM, Breiling A, Le T, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24(3):310–323.
- Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139:1895–1902. Oxford University Press for The Company of Biologists Limited
- Szwagierczak A, Bultmann S, Schmidt CS, et al. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38(19):e181.
- Muegge K. Lsh, a guardian of heterochromatin at repeat elements. Biochem Cell Biol Biochim Biol Cell. 2005;83(4):548–554.
- Dunican DS, Pennings S, Meehan RR. Lsh is essential for maintaining global DNA methylation levels in amphibia and fish and interacts directly with Dnmt1 [Internet]. Biomed Res Int. cited 2019 Jun 23]. Available from. 2015 ;2015:1–12. : https://www.hindawi.com/journals/bmri/2015/740637/.
- Han Y, Ren J, Lee E, et al. Lsh/HELLS regulates self-renewal/proliferation of neural stem/progenitor cells. Sci Rep. 2017;7(1):1136.
- Fan T, Schmidtmann A, Xi S, et al. DNA hypomethylation caused by Lsh deletion promotes erythroleukemia development. Epigenetics. 2008;3(3):134–142.
- Geiman TM, Tessarollo L, Anver MR, et al. Lsh, a SNF2 family member, is required for normal murine development. Biochim Biophys Acta BBA - Gen Subj. 2001;1526(2):211–220.
- Chronis C, Fiziev P, Papp B, et al. Cooperative binding of transcription factors orchestrates reprogramming. Cell. 2017;168(3):442–459.e20.
- Chen L, Shi Y, Liu N, et al. DNA methylation modifier LSH inhibits p53 ubiquitination and transactivates p53 to promote lipid metabolism. Epigenetics Chromatin. 2019;12(1):59.
- Zhang G, Dong Z, Prager BC, et al. Chromatin remodeler HELLS maintains glioma stem cells through E2F3 and MYC. JCI insight [Internet]. American society for clinical investigation; 2019 [ cited 2020 Sept 30];4. Available from: https://insight.jci.org/articles/view/126140
- Sun W, Zang L, Shu Q, et al. From development to diseases: the role of 5hmC in brain. Genomics. 2014;104(5):347–351.
- Kraus TFJ, Guibourt V, Kretzschmar HA. 5-Hydroxymethylcytosine, the “Sixth Base”, during brain development and ageing. J Neural Transm Vienna Austria. 1996;2015(122):1035–1043.
- Amouroux R, Nashun B, Shirane K, et al. De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat Cell Biol. 2016;18(2):225–233.
- Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Dev Camb Engl. 2012;139:1895–1902.
- Deplus R, Delatte B, Schwinn MK, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. Embo J. 2013;32(5):645–655.
- Wu H, D’Alessio AC, Ito S, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25(7):679–684.
- Ito K, Lee J, Chrysanthou S, et al. Non-catalytic roles of Tet2 are essential to regulate hematopoietic stem and progenitor cell homeostasis. Cell Rep. 2019;28(2480–2490.e4):2480–2490.e4.
- Delatte B, Deplus R, Playing FF. Playing TET ris with DNA modifications. Embo J. 2014;33(11):1198–1211.
- Han M, Li J, Cao Y, et al. A role for LSH in facilitating DNA methylation by DNMT1 through enhancing UHRF1 chromatin association. Nucleic Acids Res. 2020;48(21):12116–12134.
- Chen Q, Chen Y, Bian C, et al. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493(7433):561–564.
- Vella P, Scelfo A, Jammula S, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell. 2013;49(4):645–656.
- Imai Y, Biot M, Clément JA, et al. PRDM9 activity depends on HELLS and promotes local 5-hydroxymethylcytosine enrichment. eLife [Internet]. [ cited 2020 December 29];9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7599071/
- Pantier R, Tatar T, Colby D, et al. Endogenous epitope-tagging of Tet1, Tet2 and Tet3 identifies TET2 as a naïve pluripotency marker. Life Sci Alliance [Internet]. 2019 [cited 2020 Sept 11];2. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6776666/
- Lee DW, Zhang K, Ning ZQ, et al. Proliferation-associated SNF2-like gene (PASG): a SNF2 family member altered in leukemia. Cancer Res. 2000;60(13):3612–3622.
- Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24.
- Quivoron C, Couronné L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38.
- Velasco G, Grillo G, Touleimat N, et al. Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum Mol Genet. 2018;27(14):2409–2424.
- Unoki M, Funabiki H, Velasco G, et al. CDCA7 and HELLS mutations undermine nonhomologous end joining in centromeric instability syndrome. J Clin Invest. 2019;129(1):78–92. American Society for Clinical Investigation
- Thijssen PE, Ito Y, Grillo G, et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat Commun. 2015;6(1):7870.