
ABSTRACT
Epigenetically targeted therapeutic development, particularly for SAM-dependent methylations of DNA, mRNA and histones has been proceeding rapidly for cancer treatments over the past few years. However, this approach has barely begun to be exploited for developing new antibiotics, despite an overwhelming global need to counter antimicrobial resistance. Here, we explore whether SAM analogues, some of which are in (pre)clinical studies as inhibitors of human epigenetic enzymes, can also inhibit Clostridioides difficile-specific DNA adenine methyltransferase (CamA), a sporulation regulator present in all C. difficile genomes sequenced to date, but found in almost no other bacteria. We found that SGC0946 (an inhibitor of DOT1L), JNJ-64619178 (an inhibitor of PRMT5) and SGC8158 (an inhibitor of PRMT7) inhibit CamA enzymatic activity in vitro at low micromolar concentrations. Structural investigation of the ternary complexes of CamA-DNA in the presence of SGC0946 or SGC8158 revealed conformational rearrangements of the N-terminal arm, with no apparent disturbance of the active site. This N-terminal arm and its modulation of exchanges between SAM (the methyl donor) and SAH (the reaction product) during catalysis of methyl transfer are, to date, unique to CamA. Our work presents a substantial first step in generating potent and selective inhibitors of CamA that would serve in the near term as chemical probes to investigate the cellular mechanism(s) of CamA in controlling spore formation and colonization, and eventually as therapeutic antivirulence agents useful in treating C. difficile infection.
Introduction
Epigenetic modifications that modulate gene expression without changing the DNA sequence play fundamental roles in mammalian development, cell fate decisions, and cellular memory and plasticity [Citation1]. The environment can affect epigenetic inheritance, with implications for evolution, human health and diseases such as cancer [Citation2–4]. Even bacterially induced host epigenetic deregulations may affect host–pathogen interactions, promoting either host defences or tolerance of pathogen persistence [Citation5,Citation6], along with host cell functions involving mismatch repair, transcription regulation, and more. Among the enzyme-driven post-synthetic and post-translational epigenetic modifications, S-adenosyl-l-methionine (SAM)-dependent methylations of DNA, mRNA and histones have increasingly become therapeutic targets in cancer research [Citation7–10]. While epigenetically targeted drug development is proceeding rapidly for cancer treatment, this approach has barely begun to be exploited for developing new antibiotics, despite the overwhelming global need to counter antimicrobial resistance [Citation11,Citation12]. Furthermore, drugs with antivirulence properties, as opposed to blocking growth or viability, appear to be less likely to be selected for resistance [Citation13,Citation14]. Finally, while bacterial DNA methyltransferases (MTases) are predominantly associated with restriction-modification systems, there are a number of DNA MTases that are highly conserved among specific groups of bacteria that play important gene-regulatory roles and may serve as particularly attractive drug targets [Citation15,Citation16]. Among these conserved, regulatory DNA MTases is CamA of Clostridioides difficile (see below).
The U.S. Centers for Disease Control and Prevention has classified five antibiotic-resistant bacteria, including C. difficile, as being the most urgent threats [Citation17] (https://www.cdc.gov/drugresistance/biggest-threats.html). C. difficile infection causes life-threatening diarrhoea and a colon inflammatory disease called pseudomembranous colitis [Citation18]. C. difficile infections are seen predominantly in people who have had both recent medical care (due to in-hospital exposure) and antibiotic treatment (due to suppression of the normal colon bacteria) [Citation19]. This Gram-positive pathogen produces endospores, making decontamination difficult [Citation20,Citation21], and it causes substantial tissue damage by producing significant toxins [Citation22,Citation23]. It prolongs its presence by stabilizing the colon bacterial population (microbiota, or microbiome) in an unhealthy distribution [Citation24,Citation25]. Thus far, this critical medical need has been unmet by therapeutic strategies [Citation26–29].
Novel targeted therapeutics are urgently needed to combat C. difficile infection. First, there is an increasing resistance of C. difficile to commonly used antibiotics such as vancomycin [Citation30] and metronidazole [Citation31,Citation32]. Second, antibiotics vary in how rapidly, post treatment, they allow recovery of the C. difficile-resistant microbiome [Citation33]. A potential avenue for epigenetic-targeted treatment of C. difficile infections is provided by the recently discovered C. difficile adenine methyltransferase A (CamA), which is present in all C. difficile genomes sequenced to date (>300), but is rarely found in other bacteria [Citation34]. Importantly, CamA-mediated DNA adenine methylation at CAAAAA (underlining indicates the target A) is required for normal sporulation and biofilm production by C. difficile, key steps in the disease transmission, as well as for colonization in animal models [Citation34]. Finding the CamA inhibitors would complement ongoing searches for other antivirulence agents targeting C. difficile [Citation24] as well as providing investigative tools to characterize the currently unknown mechanism(s) by which CamA controls sporulation and biofilm production. While inhibition of sporulation can clear C. difficile infection with low recurrence [Citation35,Citation36], there has been limited discovery-oriented research on inhibitors targeting the formation of spores. Such inhibitors could be highly synergistic with antibiotics targeting the vegetative cells and, in addition, could greatly decrease the spread of the pathogen in hospital settings.
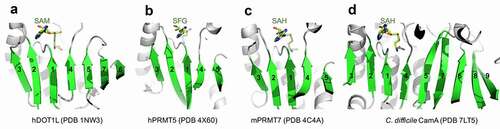
The catalytic domain of CamA [Citation37] is consistent with the Rossman-fold of Class I MTases [Citation38], which share a conserved SAM binding site (). This structure is shared by (among many other MTases) human DOT1L (a histone H3 lysine-79 MTase [Citation39]) and PRMTs (protein arginine MTases [Citation40–43]). Here, we explore whether SAM analogs, some of which are already in clinical trials as inhibitors of human epigenetic enzymes, can also inhibit CamA enzymatic activity in vitro. This would set the stage for generating potent and selective inhibitors of CamA that would serve as useful chemical probes to investigate the cellular mechanism(s) of CamA in controlling spore formation and colonization, and eventually perhaps as therapeutic antivirulence agents useful in treating C. difficile infection.
Figure 1. Examples of Class I MTases. (a) Human DOT1L. (b) Human PRMT5. (c) Human PRMT7. (d) C. difficile CamA.
Materials and methods
The highly purified recombinant proteins used in this study were all characterized previously in our laboratory: C. difficile CamA (pXC2184) [Citation37], E. coli Dam (pXC1612) [Citation44,Citation45], Caulobacter crescentus CcrM (pXC2121) [Citation46], human MettL3-14 [Citation47,Citation48], human MettL5-Trm112 (pXC2062-pXC2076), human MettL16 (pXC2210) and human PCIF1 (pXC2055) [Citation49], mouse Dnmt3a2-3L (pXC465 and pXC391) and Dnmt3b2-3L (pXC273 and pXC391) [Citation50], and human DNMT1 (pXC896) [Citation51]. Compounds of SGC epigenetic probe set (https://www.thesgc.org/chemical-probes/epigenetics) were purchased from Cayman Chemicals, with additional compounds from Selleck Chemicals, MedChemExpress, and Sigma–Aldrich.
Methylation inhibition assay
The purified CamA enzymes in 300 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) were concentrated to 2.5 mg/mL, flash-frozen in liquid nitrogen and stored in −80°C. The methylation inhibition assay of CamA was performed at room temperature (~22°C) for 2.5 min in a 20 µL reaction mixture containing 50 nM CamA, 40 µM SAM, 5 µM double-stranded DNA (5´-CGA TTC AAA AAG TCC CAA G-3´ and 3´-GCT AAG TTT TTC AGG GTT C-5´ where the underlined A is the target) and indicated concentration of inhibitor in reaction buffer of 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 0.1 mg/mL BSA, and 0.25% DMSO.
Typically, a mixture of CamA and SAM is preincubated at room temperature for 5 min, at which the same volume of DNA with or without inhibitor was added to start the reaction. The reaction was terminated by adding trifluoroacetic acid (TFA) to 0.1%.
The enzyme activity was measured by Promega luminescence assay (MTase-GloTM) [Citation52], where the produced SAH product is converted into ATP in a two-step reaction and the ATP is detected by a luciferase reaction. The 5 μL of the reaction mixture was transferred to a low-volume 384-well plate, and the luminescence signal was measured by a Synergy 4 multimode microplate reader (BioTek).
Isothermal titration calorimetry
All ITC experiments were performed using a MicroCal PEAQ-ITC automated system (Malvern) at 25°C. The reference power was set at 8 µcal/s and total of 19 injections were performed with an initial injection of 0.2 µL followed by 18 injections (each of 2 µL) with continuous stirring at 750 rpm. The spacing time between each injection was set as 300 seconds to allow equilibrium. All ITC data were processed using software supplied by the manufacturer (Malvern) and the binding constants were calculated by fitting the data as one-binding site with the offset subtracted.
For measuring CamA binding to three DOT1L inhibitors (), 900 µM inhibitor (SGC0946, EPZ004777, or EPZ5676) in the syringe was titrated into 40 µM CamA (in the cell) in 325 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.5 mM TCEP and 10% DMSO.
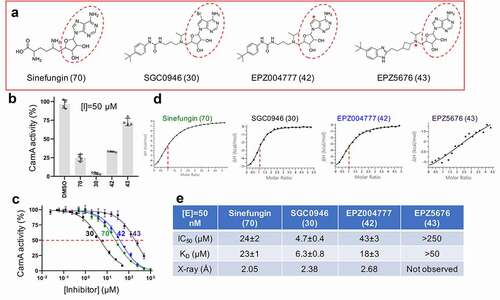
Figure 2. CamA inhibition by DOT1L inhibitors. (a) Chemical structures of sinefungin and three DOT1L inhibitors examined. The numerical number in parenthesis refers to our laboratory code. The symbol * in EPZ004777 indicates a substitution of the purine nitrogen with carbon atom. The symbol * in EPZ5676 indicates a chiral atom. (b) Relative inhibition of CamA activity at a single inhibitor concentration of 50 μM. (c) The IC50 measurements were made by varying inhibitor concentrations. The shown codes for the four inhibitors are described in panel A. (d) Isothermal titration calorimetry (ITC) measurement of dissociation constants and stoichiometry (vertical red dash line) of various compounds to CamA. (e) Summary of the IC50 and KD values of inhibition of methylation of CamA by various inhibitors on DNA substrate.
For measuring CamA binding to two PRMT inhibitors (), in order to overcome the low solubility of JNJ-64619178 and SGC8158, a reverse titration was performed with 450 µM CamA (in the syringe) titrated into 30 µM inhibitors (in the cell). The binding was performed in 500 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.5 mM TCEP and 10% DMSO.
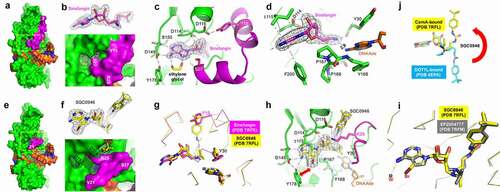
Figure 3. Structures of CamA-DNA in the presence of inhibitors. (a) Surface presentation of the ternary complex of CamA (in green and magenta)-DNA (in orange)-sinefungin (in magenta sticks). The N-terminal residue (3) is labelled. (b) The omit electron density map (contoured at 5.0 σ above the mean) for the bound sinefungin (top panel). The enlarged binding pocket of sinefungin, where the edge of adenine moiety and the ribose hydroxyl groups were visible (bottom panel). (c) An ethylene glycol binds to the edge of adenine moiety. (d) The amino group of sinefungin points to the target adenine of DNA. (e) Surface presentation of the ternary complex of CamA-DNA-SGC0946 (in yellow sticks). The first ordered N-terminal residue (21) is labelled. (f) The omit electron density map (contoured at 5.0 σ above the mean) for the bound SGC0946 (top panel). The enlarged binding pocket of SGC0946, where the bromine atom at ring position 7 of the purine edge and the terminal tri-methyl group attached to butylphenyl moiety were visible (bottom panel). (g) Superimposition of CamA with bound sinefungin (magenta) and SGC0946 (yellow) revealed that the terminal tri-methyl group of SGC0946 displaces Tyr15. (h) Rearranged Lys25–Asp116 interaction upon the binding of SGC0946. (i) Superimposition of CamA-bound SGC0946 (yellow) and EPZ004777 (grey). (j) Superimposition of CamA-bound SGC0946 (yellow) and DOT1L-bound SGC0946 (cyan).
For measuring the effect of PRMT inhibitors JNJ-64619178 and SGC8158 on CamA binding to DNA ( and Figure S4A-B), 200 µM DNA (in the syringe) was titrated to 20 µM CamA (in the cell) in the presence or absence of 100 µM inhibitors in buffer 250 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.5 mM TCEP and 0.5% DMSO.
For measuring the effect of DNA on CamA binding to cofactor (Figure S4C), 900 µM SAH or Sinefungin (in the syringe) was titrated to 40 µM CamA (in the cell) in the presence or absence of 50 µM DNA in 325 mM NaCl, 20 mM Tris-HCl pH 7.5, 0.5 mM TCEP and 10% DMSO.
X-ray crystallography
CamA-DNA complexes were initially mixed at a molar ratio of 1:1.1 and incubated on ice for 30 min and concentrated up to about 80 ~ 100 µM in buffer containing 100 mM NaCl, 20 mM Tris-HCl, and 0.5 mM TCEP. The DNA used for co-crystallization was a 14-base pair duplex (5´-TTC AAA AAG TCC CA-3´ and 3´-AGT TTT TCA GGG TA-5´). Ternary complex was made by adding 200 ~ 300 µM inhibitors (Sinefungin, SGC0946, EPZ004777, SGC8158 or JNJ-64619178) to pre-formed binary CamA-DNA complex and incubated on ice for 1 h. Sitting drops were set by an Art Robbins Gryphon Crystallization Robot by mixing 0.2 µL CamA-DNA-inhibitor complex with 0.2 µl crystallization solution with 70 µL well solution. Crystals for CamA-DNA-inhibitor ternary complex appeared under similar conditions of 21 ~ 24% (w/v) polyethylene glycol 3350, 0.1 M Tris-HCl pH 7.0 ~ 7.5, 0.28 M potassium citrate at room temperature after 3 ~ 4 days. Crystals were cryoprotected in reservoir solution supplemented with 20% (v/v) ethylene glycol and flash frozen in liquid nitrogen.
Diffraction data were collected at the SER-CAT beamline 22ID of the Advanced Photon Source at Argonne National Laboratory. The structures of the CamA-DNA-inhibitor ternary complex were solved by the difference Fourier method using the CamA-DNA structure (PDB 7LT5). Structure quality was analysed during PHENIX refinements [Citation53] together with manual inspection using Coot [Citation54]. We note that the electron density for JNJ-64619178 was not observed in a CamA-DNA complex crystal (not shown). The final structure models were validated by the PDB validation server [Citation55]. Structure images were prepared by PyMol (Schrödinger, LLC).
Results
Following the initial report of the discovery of CamA [Citation34], we have expressed and purified the recombinant CamA enzyme, optimized methylation reaction conditions, and determined its structure bound to DNA containing the recognition sequence [Citation37]. The binding by CamA of the methyl donor SAM is unexpectedly weak (KD = 35 μM and Km > 17 μM [Citation37]) compared to other DNA MTases (e.g., E. coli Dam, Km = 3–6 μM [Citation56–58]; phage T4 Dam, Km = 0.5 μM [Citation59]; and M.TaqI, KD = 2 μM [Citation60]). Furthermore, the SAM-interacting residues [Citation37] are very highly conserved among C. difficile isolates – in >300 orthologs, only five were found with substitutions affecting the relevant residues (Figure S1). This strongly suggests that the unusually weak SAM-binding by CamA is a conserved feature, and prompted us to ask whether SAM analogs are particularly potent inhibitors of CamA activity. We accordingly carried out a CamA inhibition screen, using mostly commercially available compounds developed for human epigenetic modification enzymes and modification reader domains (Figure S2). We used sinefungin, a pan inhibitor of SAM-dependent MTases, as a positive control ().
A few compounds showed notable inhibition of CamA: SGC0946 [Citation61] and EPZ004777 [Citation62] (inhibitors of histone lysine MTase DOT1L); along with JNJ-64619178 [Citation63] and SGC8158 [Citation64] (inhibitors of the protein arginine MTases PRMT5 and PRMT7). These compounds are SAM-competitive inhibitors, each containing a common adenosyl moiety and all occupying the SAM binding pocket in the respective enzymes’ active sites. Below we describe the modes of CamA inhibition by the two sets of inhibitors and discuss the differences between them.
CamA Inhibition by histone lysine MTase DOT1L inhibitors
The DOT1L inhibitors were designed initially based on the crystal structure of the DOT1L active site [Citation65], resulting in EPZ004777, which retains the nucleoside core of SAM [Citation62] (). SGC0946, a brominated analogue of EPZ004777, exhibited improved solubility and thus potency [Citation61]. We observed that the same trend between these two inhibitors against CamA: SGC0946 exhibited more potent inhibition than EPZ004777 against CamA, ~9X in IC50 values (4.7 μM vs. 43 μM; ), and stronger binding, ~3X in dissociation constants KD values (6.3 μM vs. 18 μM; -e). A related DOT1L inhibitor EPZ5676 (aka pinometostat) [Citation66] is in a clinical trial for paediatric acute myeloid leukaemia [Citation67]. However, EPZ5676 displayed much reduced inhibitory activity (IC50 > 250 μM) and much weaker binding (KD > 50 μM) to CamA.
We determined the structures of the CamA in ternary complex with sinefungin, SGC0946 or EPZ004777 in the presence of substrate DNA to resolutions of 2.05 Å, 2.38 Å and 2.68 Å, respectively (Table S1). Like the previously characterized CamA-DNA-SAH complex, the binding of the sinefungin results in observable electron density for nearly the entire N-terminal arm, starting from residue three (); that arm is unstructured in the binary CamA-DNA structure [Citation37]. Two sets of intramolecular interactions, Tyr15-Asp116 and Ser27-Ile115, close off the cofactor binding site (). An ethylene glycol, used in the cryoprotection of crystals, makes bidentate contacts with N6 and N7 of the adenosyl moiety of sinefungin (). Sinefungin carries an amino group, in the position corresponding to the transferrable methyl group of SAM, and this amino group was positioned 2.8 Å away from the target adenine N6 atom of DNA substrate ().
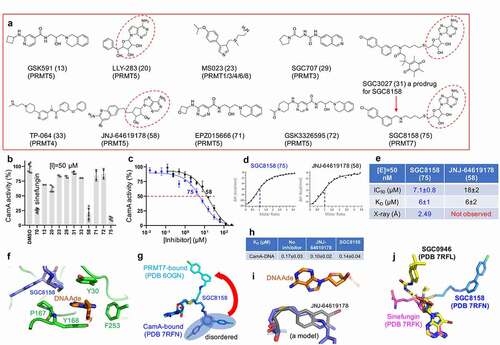
Figure 4. CamA inhibition by PRMT inhibitors. (a) Chemical structures of known PRMT inhibitors examined. The numerical number in parenthesis refers to our laboratory code. (b) Relative inhibition of CamA activity at a single inhibitor concentration of 50 μM. (c) The IC50 measurements by varying inhibitor concentrations of compounds SGC8158 and JNJ-64619178. (d) ITC measurement of dissociation constants and stoichiometry (vertical red dash line) of the two compounds to CamA. (e) Summary of the IC50 and KD values of inhibition of methylation of CamA by inhibitors SGC8158 and JNJ-64619178. (f) Structure of CamA-DNA in the presence of SGC8158, which becomes part of the binding site for the DNA target adenine. (g) Superimposition of SGC8158 with CamA-bound (blue) and PRMT7-bound (cyan). (h) The ITC dissociation constants of CamA-DNA were measured in the absence and presence of inhibitors (see Figure S4). (i) A model of JNJ-64619178 (grey) superimposed onto SGC8158 (blue), sitting next to target DNA adenine (orange). (j) Superimposition of CamA-bound sinefungin (magenta), SGC0946 (yellow) and SGC8158 (blue).
As expected, SGC0946 binds the SAM binding pocket via the adenosyl and sugar moieties, with the bulky para-tert-butylphenyl appending group extending out onto the surface (-f). The overall structure is very similar to that of CamA bound to sinefungin (root-mean-square deviation of 0.3 Å across 500 pairs of Cα atoms), with the exception of the N-terminal arm, in which residues 21–27 are structured, while residues prior to #21 remain disordered. The SGC0946 terminal tri-methyl group, attached to the butylphenyl moiety, occupies the position of Tyr15 () and disrupts the Tyr15-Asp116 interaction seen in the structure of CamA-sinefungin (), resulting in rearrangement of N-terminal residues. In addition, the bulkier dimethyl moiety pointing to the target adenine-binding pocket forces the Tyr30 aromatic ring to rotate ~90º (). The rearranged conformation of residues 21–27 involves a salt-bridge between Arg25 and Asp116 (). The bromine atom at the ring position 7 of the SGC0946 purine moiety engages in a hydrophobic interaction with Tyr178 (), which might be responsible, at least in part, for its increased potency (in both IC50 and KD values) relative to EPZ004777. The CamA structures are nearly identical when bound to EPZ004777 or SGC0946, except for a water molecule, instead of ethylene glycol, being observed near the purine N6 atom of EPZ004777 (). As observed in the structures of DOT1L [Citation61], advantages (if any) to the position 7 substitutions of the purine nitrogen with carbon in EPZ004777 were not identified in the structure with CamA. However, the derivatization of the purine 6 and 7 positions (as shown by bromination) indicate that CamA can accommodate vastly different chemical structures, presenting a possible route to optimizing binding or inhibitory activity.
Next, we superimposed two protein-bound (DOT1L or CamA) SGC0946 molecules. While the adenosyl and sugar moieties are well overlaid in the SAM binding pockets, the para-tert-butylphenyl group points in opposite directions (). The two conformations can be reached from one another by rotating the three torsion angles after the sugar ring. We note that in CamA, the t-butylphenyl end of the compound is exposed to solvent and the disordered N-terminal residues (-f), whereas in DOT1L the t-butyl group is surrounded by a cluster of hydrophobic side chains, likely contributing to its potent binding to DOT1L.
As shown in , EPZ5676 failed to inhibit CamA. We reason that the rotations of torsion angles observed with SGC0946/EPZ004777 are not applicable with the cyclobutene spacer in EPZ5676, which yields a cis/trans mixture. The cis isoform of EPZ5676 compound is separated through the chiral purification [Citation68] and is the isoform we tested.
CamA inhibition by protein arginine MTase (PRMT) inhibitors
As noted above, most members of the Class I MTases [Citation38] include a seven β-stranded catalytic domain (like DOT1L, ), whereas PRMTs contain a five stranded β-sheet (-c), and CamA contains a nine stranded β-sheet (). The past few years have witnessed a remarkable advance in the development of molecular tools and clinical compounds able to selectively and potently inhibit PRMTs [Citation69], and these act as SAM-competitive, substrate competitive or allosteric inhibitors [Citation70]. Some of them were included in our screen against CamA (). Among them, the most effective inhibitors against CamA activity were two SAM analogs, JNJ-64619178 and SGC8158 (). We note that LLY-283 is also a SAM-analog, but displayed a much weaker inhibition than sinefungin against CamA, and thus was not used further in our study. JNJ-64619178, a PRMT5 inhibitor, is in a phase-I study in adults with advanced solid tumours and non-Hodgkin's lymphoma [Citation63]. SGC8158 is a potent SAM-competitive PRMT7 inhibitor, generated from a mammalian cell permeable prodrug SGC3027 [Citation64]. As anticipated, the unprocessed prodrug SGC3027 does not possess in vitro inhibitory activity against CamA ().
While both JNJ-64619178 and SGC8158 exhibited the same binding affinity (KD = 6 μM), SGC8158 exhibited 2.5X higher inhibitory activity (-e). As expected, structural investigation revealed that SGC8158 occupies the SAM binding pocket via its adenosyl and ribose moieties. Clear electron density was observed for the methylene linker up to the biphenyl moiety. Unexpectedly, the methylene linker of SGC8158 extends towards the target adenine of the DNA substrate, becoming part of the aromatic cage-like binding site for the target adenine together with Tyr168, Tyr30 and Phe253 (). This is reminiscent of the observation that SGC8158 did not affect the peptide substrate binding by PRMT7 [Citation64]. For example, the Dot1L inhibitor analysed above, the biphenylmethylamine moiety of SGC8158 occupies a hydrophobic pocket in PRMT7 [Citation64], whereas the corresponding moiety in the CamA-bound form extends to the surface and becomes disordered (). It is encouraging to note that a derivative without the biphenyl moiety (e.g., SGC0911) still inhibits PRMT7 activity, with an IC50 of 1 μM, and its derivatization resulted in a potent PRMT7 inhibitor (e.g., SGC8158) with IC50 < 2.5 nM [Citation64]. A similar optimization approach might be applicable to CamA.
For reasons unclear to us, we did not observe JNJ-64619178 bound in the current CamA-DNA structure. We noticed a low solubility of JNJ-64619178 at 200–300 μM concentrations used for co-crystallization. We tested for JNJ-64619178 variations in solubility using samples from three different vendors, and found that they have similar potencies, inhibiting CamA activity at or below 50 μM concentrations (Figure S3). We generated a chemical model of JNJ-64619178 and superimposed it onto SGC8158. The shorter methylene linker length (3-carbon bonds) between the terminal pteridine moiety of JNJ-64619178 and the adenosine core structure could position the pteridine ring along the DNA target binding site (). We measured the dissociation constants of CamA-DNA in the presence of inhibitors, and found that both JNJ-64619178 and SGC8158 do not affect CamA-DNA binding, and may even slightly strengthen it ().
Discussion
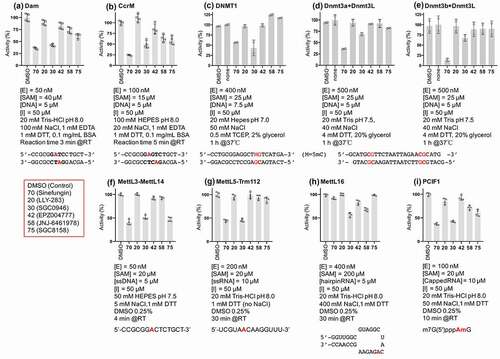
Here we present a critical first step for antivirulence drug development targeting CamA, a Clostridioides difficile-specific DNA adenine methyltransferase and sporulation regulator [Citation34]. We identified three SAM analogs (SGC0946, SGC8158 and JNJ-64619178), which are already in (pre)clinical studies as inhibitors of human epigenetic enzymes, and show that they can also inhibit CamA enzymatic activity in vitro at low micromolar concentrations. These molecules represent distinct inhibitor chemotypes (). We have been tweaking and testing compound designs, and optimizing the early lead molecules so that they behave more potently and selectively against other class I MTases, including both human and bacterial enzymes (). The DOT1L-inhibitor SGC0946 exhibits ~50% in vitro inhibition against six distinct MTase enzymes we have examined (four human RNA adenine MTases and two bacterial DNA adenine MTases), while the PRMT7-inhibitor SGC8158 has more selectivity against MettL3-MettL14, an RNA adenine MTase important in myeloid leukaemia [Citation10], which also methylates DNA in vitro [Citation47,Citation48].
Figure 5. Selectivity of compounds 30, 42, 58 and 75 (see red box) against (a-b) two bacterial DNA adenine MTases (Escherichia coli Dam and Caulobactor crescentus CcrM), (c-e) three mammalian DNA cytosine MTases (human DNMT1, mouse Dnmt3a-3L and Dnmt3b-3L), and (f-i) four human RNA adenine MTases (MettL3-MettL14, MettL5-Trm112, MettL16 and PCIF1). The reaction condition and substrate for each enzyme are indicated.
In structurally characterizing the CamA-DNA-inhibitor complexes, we found that inhibitors induce a particular kind of conformational rearrangement unique (to date) to CamA among Class I MTases. Specifically, sinefungin, SGC0946, or SGC8158 binding led to substantial movement of the CamA N-terminal arm, with no apparent disturbance of the active site. This rearrangement, originally observed with bound SAH [Citation37], results in the unstructured N-terminal arm moving to close off the SAM-binding pocket. The ability of SGC0946 and SGC8158 to induce this movement may contribute to their substantial potency as CamA inhibitors.
While outside the scope of the current work, the next step is the use of these data to identify more potent derivatives that can then be tested against C. difficile both in vitro and in a model animal infection. Although the lead compounds (or the prodrugs) are mammalian cells permeable and, in the case of JNJ-64619178, orally active, we do not know whether the molecules can efficiently cross the cell wall of Gram-positive bacteria (the group to which C. difficile belongs). This cell wall is dominated by the peptidoglycan layer, presenting a landscape of large (up to 60 nm in diameter), deep (up to 23 nm) pores constituting a disordered gel [Citation71]. Furthermore, the peptidoglycan is covered by a proteinaceous S-layer [Citation72,Citation73], and underlaid by a fairly typical lipid bilayer membrane [Citation74].
These early lead molecules provide opportunity and challenge for optimizing compound potency, selectivity, and ability to cross the cell wall of Gram-positive bacteria in particular, that of C. difficile. Finally, we note that a compound inhibiting CamA activity without killing the bacteria would represent an essential research tool for uncovering the possible mechanism(s) by which CamA epigenetically regulates gene expression for C. difficile sporulation and colonization.
Authors contributions
J.Z. performed protein purification, enzymatic assays, ITC measurements and crystallization. J.Z. and J.R.H. performed X-ray crystallography experiments. Y.D. performed assays of selectivity using human RNA adenine MTases. R.R. performed selectivity assays using human DNA cytosine MTases (DNMTs). R.M.B. performed bioinformatic analysis, participated in discussion and assisted in preparing the manuscript. X.Z and X.C organized and designed the scope of the study.
Data Availability
The experimental data that support the findings of this study are contained within the article. The X-ray structure (coordinates) and the source data (structure factor file) of CamA-DNA with bound inhibitors have been submitted to the PDB under accession numbers 7RFK (sinefungin), 7RFL (SGC0946), 7RFM (EPZ004777) and 7RFN (SGC8158).
Highlights
Conformational rearrangement of CamA N-terminal arm provides opportunities for binding distinct inhibitors
Histone lysine methyltransferase inhibitors SGC0946 and EPZ004777 bind in the SAM-binding site and each displace Tyr15 of CamA
Protein arginine methyltransferase inhibitor SGC8158, and possibly JNJ-64619178, bind in the SAM-binding site and is positioned next to the DNA target adenine
Supplemental Material
Download MS Word (1.8 MB)Acknowledgments
We thank Ms. Yu Cao for technical assistance, Mr. Sarath Pathuri for purified DNMT1, Dr. Clayton B. Woodcock for purified CcrM, Dr. Taraneh Hajian and Dr. Masoud Vedadi for purified MettL3-MettL14, and Dr. Julian Hurdle for the discussion. We thank Dr. Suzanne Ackloo at the Structural Genomics Consortium (SGC) for helping with the SGC Probe Set, which was purchased from Cayman Chemical Company.
Disclosure statement
The authors declare that no competing interests exist.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Allis CD, Jenuwein T, Reinberg D. Epigenetics, second edition (Cold Spring Harbor, New York, NY: Cold Spring Harbor Laboratory Press; 2015.
- Kaliman P. Epigenetics and meditation. Curr Opin Psychol. 2019;28:76–80.
- Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–499.
- Sapienza C, Issa JP. Diet, Nutrition, and Cancer Epigenetics. Annu Rev Nutr. 2016;36(1):665–681.
- Bierne H, Hamon M, Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2012;2(12):a010272.
- Bierne H, Hamon M. Targeting host epigenetic machinery: the Listeria paradigm. Cell Microbiol. 2020;22(4):e13169.
- Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5(1):37–50.
- Jarrold J, Davies CC. PRMTs and Arginine Methylation: cancer’s Best-Kept Secret? Trends Mol Med. 2019;25(11):993–1009.
- Siklos M, Kubicek S. Therapeutic targeting of chromatin: status and opportunities. FEBS J. 2021. DOI:10.1111/febs.15966
- Yankova E, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593(7860):597–601.
- McKenna M. The antibiotic paradox: why companies can’t afford to create life-saving drugs. Nature. 2020;584(7821):338–341.
- Plackett B. No money for new drugs. Nature. 2020;586(7830):S50–S52.
- Dickey SW, Cheung GYC, Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov. 2017;16(7):457–471.
- Ogawara H. Possible drugs for the treatment of bacterial infections in the future: anti-virulence drugs. J Antibiot (Tokyo). 2021;74(1):24–41.
- Oliveira PH, Conserved FG. DNA Methyltransferases: a Window into Fundamental Mechanisms of Epigenetic Regulation in Bacteria. Trends Microbiol. 2020;29(1):28–40.
- Anton BP, Roberts RJ. Beyond Restriction Modification: epigenomic Roles of DNA Methylation in Prokaryotes. Annu Rev Microbiol. 2021;75(1). DOI:10.1146/annurev-micro-040521-035040
- Guh AY, et al. Trends in U.S. Burden of Clostridioides difficile Infection and Outcomes. N Engl J Med. 2020;382(14):1320–1330.
- Farooq PD, Urrunaga NH, Tang DM, et al. Pseudomembranous colitis. Dis Mon. 2015;61(5):181–206.
- Webb BJ, et al. Antibiotic Exposure and Risk for Hospital-Associated Clostridioides difficile Infection. Antimicrob Agents Chemother. 2020;64(4):e02169–19.
- Rashid T, et al. Activity of Hospital Disinfectants against Vegetative Cells and Spores of Clostridioides difficile Embedded in Biofilms. Antimicrob Agents Chemother. 2019;64(1):e01031–19.
- Freedberg DE, Salmasian H, Cohen B, et al. Receipt of Antibiotics in Hospitalized Patients and Risk for Clostridium difficile Infection in Subsequent Patients Who Occupy the Same Bed. JAMA Intern Med. 2016;176(12):1801–1808.
- Markham NO, et al. Murine Intrarectal Instillation of Purified Recombinant Clostridioides difficile Toxins Enables Mechanistic Studies of Pathogenesis. Infect Immun. 2021;89(4):e00543–20.
- Skinner AM, et al. The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile. J Clin Med. 2020;10(1):96.
- Stewart D, Anwar F, Vedantam G. Anti-virulence strategies for Clostridioides difficile infection: advances and roadblocks. Gut Microbes. 2020;12(1):1802865.
- Kachrimanidou M, Tsintarakis E. Insights into the Role of Human Gut Microbiota in Clostridioides difficile Infection. Microorganisms. 2020;8(2):200.
- Gil F, Calderon IL, Fuentes JA, et al. Clostridioides (Clostridium) difficile infection: current and alternative therapeutic strategies. Future Microbiol. 2018;13(4):469–482.
- Orenstein R, Patron RL, Seville MT. Why Does Clostridium difficile Infection Recur? J Am Osteopath Assoc. 2019;119:322–326.
- Gupta A, Saha S, Khanna S. Therapies to modulate gut microbiota: past, present and future. World J Gastroenterol. 2020;26(8):777–788.
- Mondal SI, Draper LA, Ross RP, et al. Bacteriophage endolysins as a potential weapon to combat Clostridioides difficile infection. Gut Microbes. 2020;12(1):1813533.
- Saha S, et al. Increasing antibiotic resistance in Clostridioides difficile: a systematic review and meta-analysis. Anaerobe. 2019;58:35–46.
- Boekhoud IM, et al. Haem is crucial for medium-dependent metronidazole resistance in clinical isolates of Clostridioides difficile. J Antimicrob Chemother. 2021;76(7):1731–1740.
- Wu X, et al. The Integrity of Heme Is Essential For Reproducible Detection of Metronidazole-Resistant Clostridioides difficile By Agar Dilution Susceptibility Tests. J Clin Microbiol. 2021; 59(9):e00585-21.
- Lesniak NA, Schubert AM, Sinani H, et al. Clearance of Clostridioides difficile Colonization Is Associated with Antibiotic-Specific Bacterial Changes. mSphere. 2021;6(3). DOI:10.1128/mSphere.01238-20
- Oliveira PH, et al. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat Microbiol. 2020;5(1):166–180.
- Srikhanta YN, et al. Cephamycins inhibit pathogen sporulation and effectively treat recurrent Clostridioides difficile infection. Nat Microbiol. 2019;4(12):2237–2245.
- Sapkota M, Marreddy RKR, Wu X, et al. The early stage peptidoglycan biosynthesis Mur enzymes are antibacterial and antisporulation drug targets for recurrent Clostridioides difficile infection. Anaerobe. 2020;61:102129.
- Zhou J, Horton JR, Blumenthal RM, et al. Clostridioides difficile specific DNA adenine methyltransferase CamA squeezes and flips adenine out of DNA helix. Nat Commun. 2021;12(1):3436.
- Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28(6):329–335.
- Feng Q, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–1058.
- Zhang X, Zhou L, Cheng X. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. Embo J. 2000;19(14):3509–3519.
- Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11(5):509–520.
- Yue WW, Hassler M, Roe SM, et al. Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. Embo J. 2007;26(20):4402–4412.
- Sun L, et al. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc Natl Acad Sci U S A. 2011;108(51):20538–20543.
- Horton JR, Liebert K, Bekes M, et al. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J Mol Biol. 2006;358:559–570.
- Horton JR, Liebert K, Hattman S, et al. Transition from nonspecific to specific DNA interactions along the substrate-recognition pathway of dam methyltransferase. Cell. 2005;121(3):349–361.
- Horton JR, et al. The cell cycle-regulated DNA adenine methyltransferase CcrM opens a bubble at its DNA recognition site. Nat Commun. 2019;10(1):4600.
- Woodcock CB, et al. Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 2019;5(1):63.
- Yu D, et al. Human MettL3-MettL14 RNA adenine methyltransferase complex is active on double-stranded DNA containing lesions. Nucleic Acids Res. 2021. DOI:10.1093/nar/gkab460.
- Yu D, Kaur G, Blumenthal RM, et al. Enzymatic characterization of three human RNA adenosine methyltransferases reveals diverse substrate affinities and reaction optima. J Biol Chem. 2021;296:100270.
- Zeng Y, et al. The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b-mediated DNA methylation. Genes Dev. 2020;34(21–22):1546–1558.
- Hashimoto H, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40(11):4841–4849.
- Hsiao K, Zegzouti H, Goueli SA. Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics. 2016;8(3):321–339.
- Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(4):352–367.
- Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(4):486–501.
- Read RJ, et al. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19(10):1395–1412.
- Urig S, et al. The Escherichia coli dam DNA methyltransferase modifies DNA in a highly processive reaction. J Mol Biol. 2002;319(5):1085–1096.
- Bergerat A, Guschlbauer W. The double role of methyl donor and allosteric effector of S-adenosyl-methionine for Dam methylase of E. coli. Nucleic Acids Res. 1990;18(15):4369–4375.
- Marzabal S, et al. Dam methylase from Escherichia coli: kinetic studies using modified DNA oligomers: hemimethylated substrates. Nucleic Acids Res. 1995;23(18):3648–3655.
- Zinoviev VV, et al. Phage T4 DNA [N 6-Adenine] Methyltransferase: kinetic Studies Using Oligonucleotides Containing Native or Modified Recognition Sites. Biol Chem. 1998;379(4–5):481–488.
- Schluckebier G, Kozak M, Bleimling N, et al. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J Mol Biol. 1997;265(1):56–67.
- Yu W, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3(1):1288.
- Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65.
- Villar MV, et al. 537MO First-in-human study of JNJ-64619178, a protein arginine methyltransferase 5 (PRMT5) inhibitor, in patients with advanced cancers. Ann Oncol. 2020;31:S470.
- Szewczyk MM, et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat Commun. 2020;11(1):2396.
- Min J, Feng Q, Li Z, et al. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112(5):711–723.
- Daigle SR, Olhava EJ, Therkelsen CA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122(6):1017–1025.
- Lonetti A, et al. Inhibition of Methyltransferase DOT1L Sensitizes to Sorafenib Treatment AML Cells Irrespective of MLL-Rearrangements: a Novel Therapeutic Strategy for Pediatric AML. Cancers (Basel. 2020;12(7):1972.
- Lipka DB, Kuck D, Kliem C, et al. Substituted purine and 7-deazapurine compounds as modulators of epigenetic enzymes: a patent evaluation (WO2012075381. Expert Opin Ther Pat. 2013;23(4):537–543.
- Guccione E, Schwarz M, Di Tullio F, et al. Cancer synthetic vulnerabilities to protein arginine methyltransferase inhibitors. Curr Opin Pharmacol. 2021;59:33–42.
- Li ASM, et al. Chemical probes for protein arginine methyltransferases. Methods. 2020;175:30–43.
- Pasquina-Lemonche L, et al. The architecture of the Gram-positive bacterial cell wall. Nature. 2020;582(7811):294–297.
- Usenik A, et al. The CWB2 Cell Wall-Anchoring Module Is Revealed by the Crystal Structures of the Clostridium difficile Cell Wall Proteins Cwp8 and Cwp6. Structure. 2017;25(3):514–521.
- Kirk JA, et al. New class of precision antimicrobials redefines role of Clostridium difficile S-layer in virulence and viability. Sci Transl Med. 2017;9(406):eaah6813.
- Drucker DB, Wardle HM, Boote V. Phospholipid profiles of Clostridium difficile. J Bacteriol. 1996;178(19):5844–5846.