
ABSTRACT
Recent studies have unveiled an association between an L61R substitution within the human histone H3.3 protein and the presentation of neurodevelopmental disorders in two patients. In both cases, the mutation responsible for this substitution is encoded by one allele of the H3F3A gene and, if this mutation is indeed responsible for the disease phenotypes, it must act in a dominant fashion since the genomes of these patients also harbour three other alleles encoding wild-type histone H3.3. In our previous work in yeast, we have shown that most amino acid substitutions at H3-L61 cause an accumulation of the Spt16 component of the yFACT histone chaperone complex at the 3’ end of transcribed genes, a defect we have attributed to impaired yFACT dissociation from chromatin following transcription. In those studies, however, the H3-L61R mutant had not been tested since it does not sustain viability when expressed as the sole source of histone H3 in cells. In the present work, we show that H3-L61R impairs proper Spt16 dissociation from genes when co-expressed with wild-type histone H3 in haploid cells as well as in diploid cells that express the mutant protein from one of four histone H3-encoding alleles. These results, combined with other studies linking loss of function mutations in human Spt16 and neurodevelopmental disorders, provide a possible molecular mechanism underlying the neurodevelopmental disorders seen in patients expressing the histone H3.3 L61R mutant.
Introduction
The first level of compaction of eukaryotic chromosomes is achieved through the formation of nucleosomes, structures composed of ~147 base pairs of DNA wrapped around histone octamers, themselves composed of pairs of the core histones H2A, H2B, H3 and H4 [Citation1]. Nucleosomes are highly dynamic and play key regulatory roles in essentially all chromatin-based processes, including DNA replication, repair, and transcription. The human genome expresses several histone H3 isoforms, including the canonical H3.1 and H3.2 proteins and the closely related H3.3 variant – whereas H3.1 and H3.2 are expressed exclusively during S-phase and are incorporated into chromosomes during DNA replication, H3.3 is constitutively expressed and incorporates onto DNA through replication-independent routes (reviewed in [Citation2,Citation3]). H3.3 is found in association with highly transcribed genes as well as some heterochromatic regions (reviewed in [Citation4]).
In human cells, histone H3.3 is encoded by two genes, H3F3A and H3F3B, each controlled by distinct regulatory sequences. Strong evidence has accumulated in recent years for a causative role of specific mutant versions of H3.3 in several human cancers, including paediatric gliomas, chondroblastoma, and osteosarcomas. These mutations are somatic in nature and are found in both H3F3A and H3F3B, with the most common mutations identified so far resulting in H3.3 mutants bearing the K27M, G34R/V/W, or K36M substitutions, which, directly or indirectly, cause alterations in H3.3 posttranslation modifications (reviewed in [Citation3,Citation5]). A large-scale survey has identified many other histone mutants with possible connections to other cancers [Citation6], thus pointing to a potentially extensive impact of histone mutants on cancer biology.
Recent work has identified de novo germline mutations in H3F3A and H3F3B, but rather than being correlated with cancer, these mutations are associated with neurodevelopmental disorders. In one study, 37 distinct mutations across H3F3A and H3F3B were identified among 46 patients displaying various degrees of developmental delays and congenital anomalies [Citation7]. In each of these cases, a single allele of either H3F3A and H3F3B was identified as being mutant, thus suggesting that these mutations are dominant in nature and that the resulting mutant proteins confer disease phenotypes when present at roughly 25% of total H3.3 protein level within cells. One of the mutations found at H3F3A is a T to G transversion at position 185, resulting in a L61R amino acid substitution in the encoded H3.3 protein. Interestingly, an independent study identified the same mutation (185 T > G, H3.3 L61R) in a different patient also displaying various types of developmental disorders ([Citation8] – we note that the authors of this latter study refer to this mutant as L62R reflecting an alternative residue numbering system that includes the initiator methionine in H3.3 that is normally excised from the mature protein). Collectively, these findings thus point to germline mutations in H3.3-encoding genes as potentially being causative for neurodevelopmental disorders in humans, with the H3.3 L61R mutant being of particular interest given its identification in two independent studies.
Aside from a centromere-specific histone H3 variant, the budding yeast Saccharomyces cerevisiae expresses a single histone H3 protein that is most closely related to the human H3.3 isoform, the two of them sharing 90% amino acid identity with each other [Citation9]. In our previous work, we demonstrated that single amino substitutions at the H3-L61 position cause a shift in the distribution of the yeast FACT (yFACT, FAcilitates Chromatin Transcription/Transactions) histone chaperone complex towards the 3’ end of transcribed genes, a defect likely reflective of impaired dissociation of yFACT from genes following transcription [Citation10,Citation11]. yFACT is composed of two proteins, Spt16 and Pob3, which, through the assistance of the DNA architectural protein Nhp6, interact and reorganize nucleosomes in order to facilitate a variety of chromatin-based processes, including DNA transcription, replication, and repair (reviewed in [Citation12–16]). During transcription elongation in vivo, FACT’s major role appears to be in promoting the reassembly of nucleosomes in the wake of RNA Polymerase II (Pol II) passage in order to maintain chromatin integrity across genes, but in vitro studies, including those that originally identified FACT, have also shown a role for FACT in facilitating the disassembly of nucleosomes in order to allow Pol II access to the template DNA ([Citation17] and reviewed in [Citation13,Citation15]). Recent work has provided strong evidence for a model in which yFACT is first recruited to genes through interactions with +1 nucleosomes that have been partially unwrapped by an engaged Pol II and subsequently travels towards their 3’ ends through a processive mechanism that depends on the activity of the chromatin remodeler Chd1 [Citation18,Citation19].
The H3-L61 residue is located within a nucleosomal region we have termed ISGI (Influences Spt16-Gene Interactions), the integrity of which is required for proper physical interactions between Spt16 and transcribed genes [Citation20]. In a study in which all possible amino acid substitutions at H3-L61 were generated, we found that whereas sixteen H3-L61 mutants supported viability when the respective mutant proteins were expressed as sole source of histone H3 in cells, all sixteen caused varying degrees of defects in Spt16-gene interactions [Citation11]. The remaining three mutants, which included H3-L61R, were shown to be unable to support cell growth in the absence of wild-type H3 and were therefore not tested for defects in Spt16-gene interactions. In the present study, we carried out further analyses of the H3-L61R mutant and found that whereas it allows for cell viability when wild-type histone H3 is co-expressed in cells, it also confers strong dominant defects on Spt16-gene interactions. Our findings, combined with another recent study implicating mutant versions of the human Spt16 protein in neurodevelopmental disorders [Citation21], point to a possible molecular mechanism driving disease state in individuals expressing H3.3 L61R.
Materials and methods
Yeast strains and growth media
All strains used in this study are GAL2+ derivatives of the S288C strain background [Citation22] and are shown in . The generation of the HHT2 alleles encoding the H3-L61A, -L61W, and -L61R mutants has been described elsewhere [Citation11]. Details on standard yeast genetic techniques and media preparation have been provided previously [Citation23].
Table 1. Saccharomyces cerevisiae strains.
Chromatin immunoprecipitation (ChIP)/qPCR assays
ChIP assays were carried out using rabbit polyclonal antibodies specific for Spt16 (kindly provided by Tim Formosa) as detailed in previous studies [Citation24]. qPCRs were conducted using the following primer sets: 5'PMA1, OAD394 and OAD395; 3'PMA1, OAD383 and OAD384; 5’ FBA1, OAD419 and OAD420; 3'FBA1, OAD423 and OAD424; 5'ADH1, OAD701 and OAD702; 3'ADH1, OAD703 and OAD704. The coordinates for these primers have been previously reported [Citation25]. The sequences for the PMA1 and FBA1 primers have been provided in earlier work [Citation24,Citation26], and the sequences for the ADH1 primers are: OAD701 5’ AAAGCCAAAGGCCAACGAA 3’; OAD702 5’ AGCGTGCAAGTCAGTGTGACA 3’; OAD703 5’ TCGGTGTGTATTTTATGTCCTCAGA 3’; OAD704 5’ CTGTGGATCCGTGTGGAAGA 3’.
Results and discussion
Most H3-L61 mutant proteins support cell viability and cause varying degrees of Spt16-gene interaction defects when expressed as sole source of histone H3 in yeast cells, with the two most severe mutants, H3-L61K and H3-L61W, also causing strong growth defects [Citation11]. The H3-L61R mutant is unable to sustain viability on its own [Citation11], and we reasoned that a possible reason for this could be that it causes perturbations in Spt16 association across genes that are more pronounced than those seen in the context of the strongest viable H3-L61 mutants, and that these are extreme enough to cause lethality. To investigate this possibility, we analysed the effects of H3-L61R on Spt16-gene interactions in cells that also express wild-type histone H3.
The haploid yeast genome harbours two genes expressing histone H3, HHT1 and HHT2. In our previous work, in order to test the effects of H3 mutants in the absence of wild-type histone H3, we carried out experiments in haploid cells deleted for HHT1 and carrying either wild-type or mutant HHT2 genes (hht1∆;HHT2 and hht1∆;hht2 cells – note that in order to maintain proper histones H3 and H4 stoichiometry the HHF1 gene was also deleted in these cells [Citation11]). In the present study, we carried out experiments in haploid cells carrying wild-type HHT1 and either wild-type or mutant HHT2 genes (HHT1;HHT2 and HHT1;hht2 cells).
To assess the effect of H3-L61R on Spt16-gene interactions, we performed chromatin immunoprecipitation (ChIP) assays directed against Spt16 across three highly transcribed genes, PMA1, FBA1, and ADH1. Whereas in wild-type cells Spt16 binding is lower at the 3’ regions of these genes with respect to their corresponding 5’ regions (reflecting proper Spt16 dissociation at the end of the transcription process), cells co-expressing wild-type H3 and H3-L61R (H3 WT/H3-L61R cells) showed a marked increase in relative Spt16 occupancy at the 3’ regions of all three genes (). We tested two additional H3-L61 mutants in these assays, H3-L61A and H3-L61W – both of these mutants were previously shown to support viability when expressed as sole source of histone H3 in cells, with H3-L61A causing moderate defects in Spt16-gene interactions and H3-L61W causing the strongest defects among all viable H3-L61 mutants [Citation11]. As shown in , H3 WT/H3-L61A cells showed at most a subtle defect in Spt16 occupancy across the genes tested while H3 WT/H3-L61W cells showed much stronger defects, but lower in magnitude than those seen in H3 WT/H3-L61R cells. The defects seen in Spt16-gene interactions in H3 WT/H3 mutant cells are due to gain-of-function effects since cells deleted for HHT2 (HHT1;hht2∆, or H3 WT/H3∆ cells) behaved indistinguishably from wild-type cells in these assays (). These results demonstrate that the H3-L61R mutant causes an abnormal increase in Spt16 occupancy at the 3’ ends of transcribed genes relative to their respective 5’ ends through a dominant, gain-of-function mechanism.
Figure 1. Effects of histone H3-L61R and two other H3-L61 mutants on Spt16 occupancy across three transcribed genes when expressed in conjunction with wild-type histone H3. (a) Diagrams of the three genes tested in these assays, shown roughly to scale. Numbers above the genes represent base-pair positions starting with the first base-pair of the respective open reading frame analysed. The numbers and coloured bars below the diagrams indicate the locations of the 5’ and 3’ regions amplified in qPCR experiments. Arrows indicate the direction of transcription. (b) Levels of Spt16 occupancy across the PMA1, FBA1, ADH1 genes as measured in ChIP/qPCR assays in strains expressing the indicated histone H3 proteins. All strains express wild-type histone H3 from the HHT1 locus and either wild-type histone H3 or the indicated H3-L61 mutant from the HHT2 locus – the H3WT/H3∆ control strain is deleted for the HHT2 locus. For each gene tested, Spt16 occupancy level at the 3’ region is shown relative to Spt16 binding at the respective 5’ end (see for Spt16 % immunoprecipitation values at the 5’ regions). In each case, data are presented as mean ± S.E.M. from three independent experiments. Statistically significant differences relative to H3 WT/H3 WT cells are indicated by asterisks (Student’s t-tests, P < 0.05). The strains used in these experiments are yADP127-yADP131.
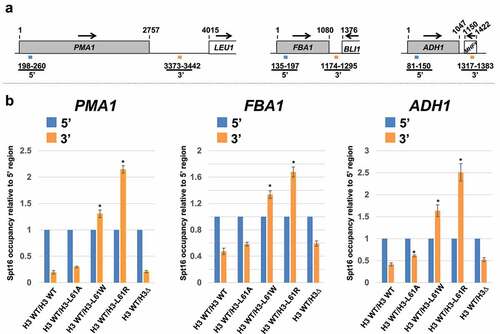
Table 2. Average Spt16 % immunoprecipitation (%IP) values at 5’ regions of genes assayed in this study.
The data in also show that H3-L61R confers stronger defects than those caused by H3-L61W, which may at least in part explain why cells expressing H3-L61R as only source of histone H3 are unable to proliferate. The other two lethal histone H3 mutants, H3-L61D and H3-L61P, also confer dominant defects in Spt16 gene dissociation, but do so to a degree comparable (H3-L61D) or less than (H3-L61P) than what is seen in H3 WT/H3-L61W cells (Figure S1), indicating that their lethal phenotypes may be due to impairment of additional processes. The defects caused by H3-L61R, -L61D, and -L61P are likely to be more widespread than the genes we tested here and are at least in part specific to Spt16 since our previous work has shown that both H3-L61W and another strong ISGI mutant, H4-R36A, cause severe Spt16 dissociation defects at several additional genes and they only confer relatively minor defects on Pol II and nucleosome occupancy across genes [Citation10,Citation26].
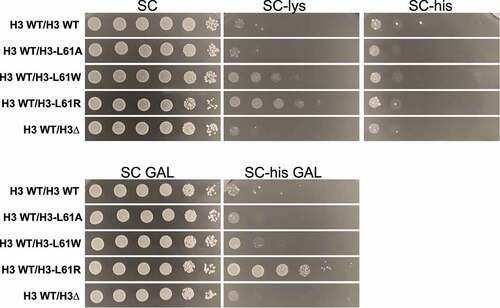
We next carried out growth tests to determine if H3-L61R confers additional dominant defects. In one experiment, we tested for the ability of the H3-L61R mutant to suppress the Lys− phenotype caused by the presence of a Ty1 δ element within the promoter of the LYS2 gene. Suppression of this allele (known as lys2-128δ) is caused by mutations that alter chromatin structure and/or the transcription machinery in a way that allows for activation of the otherwise inactive Ty1 δ promoter – the resulting phenotype, referred to as Spt−, can then be followed by growth on media lacking lysine [Citation27]. In a second experiment, we assessed if H3-L61R causes a phenotype indicative of cryptic intragenic transcription initiation. Previous work has shown that certain mutations that impair transcription-dependent nucleosome reassembly or otherwise affect chromatin environments can result in intragenic transcription initiation, and that these defects can be followed through the use of a reporter system consisting of the HIS3 coding region fused downstream from a cryptic promoter element present within the FLO8 coding sequence and assessing for the ability of his3∆ cells to grow on media lacking histidine [Citation28].
As shown in , whereas H3 WT/H3 WT cells were unable to grow on media lacking either lysine or histidine, H3 WT/H3-L61R cells displayed some growth on media lacking lysine (Spt− phenotype) and media lacking histidine (reflecting intragenic transcription initiation at the FLO8-HIS3 reporter gene), thus suggesting that the H3-L61R mutant imparts defects on chromatin structure and transcription in a dominant fashion (see ). H3 WT/H3-L61W cells also displayed an Spt− phenotype, but weaker than that seen in H3 WT/H3-L61R cells, and WT/H3-L61A cells behave similarly to wild-type cells on all media (). The fact that H3 WT/H3∆ cells behave like wild-type cells in these assays () indicates that H3-L61R perturbs chromatin structure and transcription through a gain of function mechanism.
Figure 2. Growth tests assessing dominant effects of H3-L61 mutants on phenotypes indicative of chromatin and transcription defects. Cells expressing the indicated histone H3 proteins were spotted on the indicated growth media in a 10-fold dilution series (6 μl per spot, with the cell concentration of the most concentrated spots at 5 × 108 cells/ml). Media in the upper row contain glucose, whereas those on the bottom contain galactose – the FL08:HIS3 fusion gene is under the control of the GAL1 promoter, and cryptic transcription intragenic transcription initiation in H3 WT/H3-L61R cells is only seen in conditions that activate expression of the fusion gene (i.e., in the presence of galactose). Plates were incubated at 30°C for 2 days (SC), 3 days (SC-lys, SC GAL, and SC-his GAL), or 4 days (SC-his). The strains used for these assays are yADP134-138.
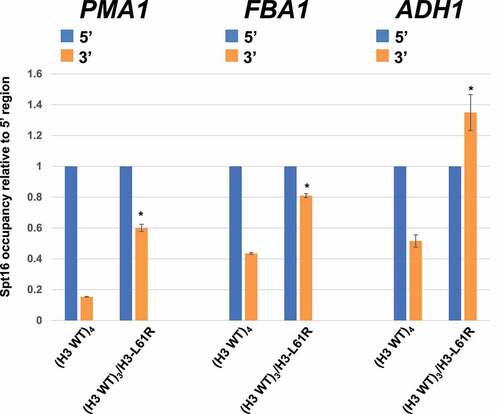
Whereas the experiments shown in demonstrate that H3-L61R can interfere with normal Spt16-gene interactions in a dominant fashion, they may not accurately reflect what occurs in human cells since the cells used for those experiments were haploid and carry a total of two histone H3-encoding alleles. To more closely mirror the histone H3.3 genetics in human cells, we generated diploid yeast cells with either a full set of wild-type histone H3-encoding alleles (HHT1/HHT1;HHT2/HHT2, or (H3 WT)4 cells) or cells with one mutant HHT2 allele (HHT1/HHT1;HHT2/hht2, or (H3 WT)3/H3-L61R cells) and assayed them in Spt16-ChIP experiments. As shown in , compared to (H3 WT)4 cells, (H3 WT)3/H3-L61R cells display increased Spt16 occupancy at the 3’ regions of all three genes tested relative to their respective 5’ regions. These results show that even when present at ~25% of total histone H3 levels in cells, the H3-L61R mutant can cause significant perturbations in Spt16-gene interactions.
Figure 3. Effects of histone H3-L61R on Spt16-gene interactions when expressed from one of four histone H3-encoding alleles in diploid cells. Spt16 occupancy in diploid cells harbouring either a full set of wild-type histone H3-encoding alleles ((H3 WT)4) or three wild-type alleles and one encoding H3-L61R ((H3 WT)3/H3-L61R)) was assessed by ChIP/qPCR at the same regions as those assayed in . For each gene tested, Spt16 occupancy level at the 3’ region is shown relative to Spt16 binding at the respective 5’ end (see for Spt16 % immunoprecipitation values at the 5’ regions). In each case, data are presented as mean ± S.E.M. from three independent experiments. Statistically significant differences relative to (H3 WT)4 cells are indicated by asterisks (Student’s t-tests, P < 0.05). The strains used in these experiments are yADP132 and yADP133.
Our results demonstrate that in yeast H3-L61R causes defects in association of Spt16 across transcribed genes through a dominant, gain of function mechanism, even when expressed in diploid cells from one of the four histone H3-encoding alleles. It is therefore possible that human cells expressing the H3.3 L61R mutant from one of the four H3.3-encoding alleles may also display similar defects in Spt16-gene interactions. Previous studies have established an association between five de novo mutations in SUPT16H, the gene encoding human Spt16, and neurodevelopmental disorders similar to those seen in the two patients carrying the H3F3A 185 T > G (H3.3 L61R) mutation [Citation21]. Whereas four of these SUPT16H mutations are point mutations resulting in single amino acid substitutions across the Spt16 protein, one is a chromosomal deletion that removes one of the two SUPT16H alleles, thus suggesting that these mutations mediate their effects through a loss-of-function mechanism. Combined with these findings, our results raise the possibility that H3.3. L61R causes neurodevelopmental disorders by trapping Spt16 at the 3’ ends of genes, effectively lowering the net amount of Spt16 available to carry out critical chromosomal functions.
The H3-L61 residue is located near other residues on the nucleosome that when mutated also cause defects in Spt16 dissociation at the 3’ end of transcribed genes, defining a nucleosomal region we have termed ISGI (Influences Spt16-Gene Interactions [Citation20]). The mechanism by which ISGI mutants impair Spt16-gene dissociation is currently unknown, but given results from structural studies showing close proximity between the ISGI region and the Spt16 middle domain [Citation29–31] it is possible that this region promotes FACT dissociation through dynamic physical interactions with Spt16, and that specific ISGI mutants perturb these interactions to a degree that they interfere with this process. Such a model would predict that different substitutions at H3-L61 would impact the ISGI region structure to different degrees and would therefore have varying effects on Spt16 dissociation, which is in line with this and our previous work [Citation11]. In future studies, it will be of interest to determine if other amino acid substitutions within the ISGI region also show an association with neurodevelopmental conditions in humans. Large-scale studies assessing the effects of H3-L61 mutants on Spt16 interactions across an entire genome will also provide important insights into the overall scope of the defects conferred by this class of histone mutants on Spt16-chromatin interactions.
Author contributions
APK, MGT, and GAT carried out the experiments related to the results shown in . APK also performed the experiments related to the results shown in . EGD, AGO and CLT performed the experiments shown in Figure S1. AAD designed and supervised all experiments and wrote the manuscript. All authors have read this paper and agree with the content.
Supplemental Material
Download MS Power Point (54.9 KB)Acknowledgments
The authors thank Tim Formosa for providing the Spt16-specific antibodies used in the ChIP assays and Reine Protacio for valuable comments during the preparation of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2022.2121073
Additional information
Funding
Related Research Data
References
- Luger K, Mader AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260.
- Shi L, Wen H, Shi X. The histone variant H3.3 in transcriptional regulation and human disease. J Mol Biol. 2017;429(13):1934–1945.
- Kumar VC, Pai R. Genes of the month: H3.3 histone genes: H3F3A and H3F3B. J Clin Pathol. 2021;74(12):753–758.
- Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21(3):421–434.
- Lowe BR, Maxham LA, Hamey JJ, et al. Histone H3 mutations: an updated view of their role in chromatin deregulation and cancer. Cancers (Basel). 2019;11(5):660.
- Nacev BA, Feng L, Bagert JD, et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature. 2019;567:473–478.
- Bryant L, Li D, Cox SG, et al. Histone H3.3 beyond cancer: germline mutations in Histone 3 Family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients. Sci Adv. 2020;6(49). DOI:10.1126/sciadv.abc9207
- Maver A, Cuturilo G, Ruml SJ, et al. Clinical next generation sequencing reveals an H3F3A gene as a new potential gene candidate for microcephaly associated with severe developmental delay, intellectual disability and growth retardation. Balkan J Med Genet. 2019;22(2):65–68.
- McBurney KL, Leung A, Choi JK, et al. Divergent residues within histone H3 dictate a unique chromatin structure in Saccharomyces cerevisiae. Genetics. 2016;202(1):341–349.
- Duina AA, Rufiange A, Bracey J, et al. Evidence that the localization of the elongation factor Spt16 across transcribed genes is dependent upon histone H3 integrity in Saccharomyces cerevisiae. Genetics. 2007;177(1):101–112.
- Johnson P, Mitchell V, McClure K, et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 2015;5(5):741–749.
- Formosa T, Winston F. The role of FACT in managing chromatin: disruption, assembly, or repair? Nucleic Acids Res. 2020;48(21):11929–11941.
- Gurova K, Chang HW, Valieva ME, et al. Structure and function of the histone chaperone FACT - Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech. 2018;1861(9):892–904.
- Bhakat KK, Ray S. The facilitates chromatin transcription (FACT) complex: its roles in DNA repair and implications for cancer therapy. DNA Repair (Amst). 2022;109:103246.
- Jeronimo C, Robert F. The histone chaperone FACT: a guardian of chromatin structure integrity. Transcription. 2022;1–23. DOI:10.1080/21541264.2022.2069995
- Zhou K, Liu Y, Luger K. Histone chaperone FACT facilitates chromatin transcription: mechanistic and structural insights. Curr Opin Struct Biol. 2020;65:26–32.
- Orphanides G, LeRoy G, Chang CH, et al. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell. 1998;92(1):105–116.
- Jeronimo C, Angel A, Nguyen VQ, et al. FACT is recruited to the +1 nucleosome of transcribed genes and spreads in a Chd1-dependent manner. Mol Cell. 2021;81(17):3542–3559 e3511.
- Martin BJE, Chruscicki AT, Howe LJ. Transcription promotes the interaction of the facilitates chromatin transactions (FACT) complex with nucleosomes in Saccharomyces cerevisiae. Genetics. 2018;210(3):869–881.
- Nyamugenda E, Cox AB, Pierce JB, et al. Charged residues on the side of the nucleosome contribute to normal Spt16-gene interactions in budding yeast. Epigenetics. 2018;13(1):1–7.
- Bina R, Matalon D, Fregeau B, et al. De novo variants in SUPT16H cause neurodevelopmental disorders associated with corpus callosum abnormalities. J Med Genet. 2020;57(7):461–465.
- Winston F, Dollard C, Ricupero-Hovasse SL. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast. 1995;11(1):53–55.
- Rose MD, Winston F, Hieter P. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor NY: Cold Spring Harbor Laboratory Press; 1990.
- Myers CN, Berner GB, Holthoff JH, et al. Mutant versions of the S. cerevisiae transcription elongation factor Spt16 define regions of Spt16 that functionally interact with histone H3. PLoS ONE. 2011;6(6):e20847.
- Campbell JB, Edwards MJ, Ozersky SA, et al. Evidence that dissociation of Spt16 from transcribed genes is partially dependent on RNA Polymerase II termination. Transcription. 2019;10(4–5):195–206.
- Nguyen HT, Wharton Ws2nd, Harper JA, et al. A nucleosomal region important for ensuring proper interactions between the transcription elongation factor Spt16 and transcribed genes in Saccharomyces cerevisiae. G3 (Bethesda). 2013;3(6):929–940.
- Winston F. Transcription Regulation. NY: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 1992. p. 1271–1293.
- Cheung V, Chua G, Batada NN, et al. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008;6(11):e277.
- Tsunaka Y, Fujiwara Y, Oyama T, et al. Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev. 2016;30(6):673–686.
- Liu Y, Zhou K, Zhang N, et al. FACT caught in the act of manipulating the nucleosome. Nature. 2020;577(7790):426–431.
- Sivkina AL, Karlova MG, Valieva ME, et al. Electron microscopy analysis of ATP-independent nucleosome unfolding by FACT. Commun Biol. 2022;5(2). DOI:10.1038/s42003-021-02948-8