
ABSTRACT
Severe acne is a chronic inflammatory skin condition that is affected by both genetic and environmental factors. DNA methylation is associated with a variety of inflammatory skin diseases, but its role in severe acne is unclear. In this study, we conducted a two-stage epigenome correlation study using 88 blood samples to identify disease-related differential methylation sites. We found close associations between the DNA methylation at 23 differentially methylated sites (DMSs) and severe acne, including PDGFD, ARHGEF10, etc. Further analysis revealed that differentially methylated genes (PARP8 and MAPKAPK2) were also expressed differently between severe acne and health control groups. These findings lead us to speculation that epigenetic mechanisms may play an important role in the pathogenesis of severe acne.
KEYWORDS:
Introduction
Acne vulgaris is the most common inflammatory skin disease involving sebaceous follicles in adolescents [Citation1,Citation2]. Severe acne, the most severe form of acne [Citation3], has a wide range of skin lesions, as well as potential scars, which cause psychological harm to the patient [Citation4]. The pathogenesis of acne vulgaris is incompletely understood, but multiple studies have shown that hereditary factors play an important role in acne vulgaris, especially in severe acne [Citation5–7].
DNA methylation at the fifth position of cytosine (5mC) is a reversible genetic epigenetic process that regulates gene expression [Citation8]. Methylation can influence the binding of transcription factors to gene regulatory elements. In addition, sequence changes are closely related to DNA methylation. This view is supported by some evidence in human tissues and cultured cells [Citation9]. DNA methylation is involved in the pathogeneses of several immune skin diseases, such as psoriasis [Citation10,Citation11] and systemic lupus erythematosus (SLE) [Citation12], and other inflammatory skin diseases [Citation13]. Inflammation plays an important role in the progression of acne lesions, and presents throughout all stages of acne [Citation14]. However, to our knowledge, genomic DNA methylation changes in severe acne have not been reported.
To analyse the DNA methylation differences in severe acne, we conducted two stages of genome-wide methylation study of severe acne on 44 patients and 44 health controls using Infinium Human-Methylation EPIC array. The EPIC chip, which shows high reproducibility, interrogates more than 850,000 genomic loci with the single nucleotide resolution and contains over 90% of 450 array probes [Citation15]. To identify differentially methylated genes that also differ in gene expression in severe acne, we performed transcriptome sequencing on five severe acne patients and four health controls blood samples.
Material and methods
Study population
We performed 2 stages of epigenome-wide association analysis. There were 48 samples in stage I, 24 controls and 24 patients each. Stage II consisted of 40 samples, including 20 controls and 20 patients. Demographics of the 88 samples were shown in Supplementary Table S1. In addition, 5 patients and 4 healthy controls were included for differential expression analysis (Supplementary Table S2). The blood samples were collected from the Department of Dermatology, First Affiliated Hospital of Kunming Medical University. All the participants were from the Yunnan province of China. The recruited patients with severe acne met the Grade 4 of Pillsbury classification and did not receive systemic anti-acne treatment for 2 weeks before blood sampling. Also, the patients had not used other drugs within 2 weeks to avoid the effect of drugs on methylation. The dermatologists who collected the samples had standard diagnostic and grading training. Health controls were acquired from normal volunteers who matched the age and gender of patients with severe acne and had no acne or other androgen-related disorders or systemic genetic disorders. Health controls had not used any drugs within 2 weeks. Methylation levels are influenced by multiple factors, including gender and age. Therefore, we limited the age of the enrolled subjects to the range of 18 to 26 years and selected only male subjects. The age range of subjects for RNA-seq was 14 to 26 years, and only male subjects were selected. Written informed consent was obtained from all the subjects, and this study was permitted by the medical ethics committee of Dali University (MECDU-201801-12).
DNA methylation
A standard DNA extraction method was used to isolate DNA from peripheral blood mononuclear cells. 500 ng genomic DNA was used for bisulphite conversion. Then bisulphite converted DNA was processed using the commercial Infinium Methylation EPIC BeadChip, following the manufacturer’s protocol. 866, 836 CpG sites are contained in the EPIC. GenomeStudio software was used to transform EPIC intensity signals into idat files. Next, the idat files were processed using ChAMP package for quality control [Citation16]. The quality control included the removement of: (i) probe with detection P > 0.01, (ii) probe with beadcount<3 in 5% samples, (iii) no CpG site, (iv) SNP site, (v) multihit site. The signal intensities for the unmethylated and methylated status were standardized using the beta-mixture quantile normalization (BMIQ) algorithm [Citation17].
Differential analysis of peripheral blood cells
We quantified the cellular composition highlighting differences between severe acne and control. ChAMP’s champ.refbase function was used to calculate blood-cell proportions using a reference-based method [Citation18]. A total of 6 major blood cell types were counted from all samples: NK cells, CD4+ T cells, CD8+ T cells, B cells, granulocytes, and monocytes.
Epigenome-wide association analysis
We performed 2 stages of epigenome-wide association analysis. There were 48 samples in stage I, 24 controls and 24 patients each. Stage II consisted of 40 samples, including 20 controls and 20 patients. Across every methylation site, the level of methylation was recorded as a β value ranging from 0 (unmethylated) to 1 (methylated). By using champ.runCombat, batch effects, such as array position and slide, were removed. Principal component analysis (PCA) was conducted to detect the differences between samples based on the methylation profiles. Using a reference-based method in the champ.refbase function, the effect of blood-cell proportion was removed by adjusting the β-values matrix. The linear model adjusted for age was used to identify severe acne-related differentially methylated sites (DMSs) by limma package. Probe methylation was considered significantly different between patients and controls at P < 0.05 and a minimum methylation difference of 0.05 between groups (|delta β|≥ 0.05).
Significance of overlapping DMSs between the two stages was tested using hypergeometric test. The meta-analysis was conducted based on results from the two stages of genome-wide methylation studies and used of commonly methylation probes. Meta-analysis was performed with a fixed effect model (low heterogeneity) or random effects model (high heterogeneity), weighted by the corresponding inversed variance, which was implemented by metagen function within the Meta package [Citation19]. Using Cochran’s Q test, sites were considered significantly heterogeneous if the P-value (corrected for the number of significant sites) was less than 0.05. False discovery rates (FDR) were computed and Position where FDR<0.05 was considered genome-wide significant.
Sensitivity analyses of epigenome-wide association
In order to assess whether the epigenome-wide association results were robust to different analyses strategies, we conducted various sensitivity analyses. We performed an epigenome-wide association analysis on M-values (defined as log2(β/(1-β)) instead of β-values. Probe methylation was considered significantly different between patients and controls at P < 0.05 in both 2 stages. FDR<0.05 was considered genome-wide significant in meta-analysed result. To compare the 2 analysis strategies, we adapted a quantile-quantile (QQ) plot.
Identify severe acne-related differential expression genes (DEGs)
RNA was extracted from blood samples of 5 patients and 4 healthy controls by RNeasy Kits according to standard protocols. RNA was fragmented, adapter ligated, and fragment amplified according to Illumina protocol. The prepared libraries were sequenced on an Illumina Novaseq platform and generated 150bp paired-end reads. The software Fastqc was used to do quality control (QC) check for RNA-seq data, and the software MultiQC was used to integrate QC reports for all samples [Citation20]. Then we used the Trim Galore to trim low-quality reads and sequencing adapters. The length parameter, specifying the minimum threshold in base pairs for retaining a sequence, was set to 75. The obtained clean reads were aligned to the reference sequence (GRCH38) using the software Hisat2 with the default parameters [Citation21]. Uniquely mapped reads were used to calculate the number of reads mapped to each gene by FeatureCounts [Citation22]. Differentially expressed genes were analysed by DEseq2 package in R [Citation23], and the conditions were set as adjusted P < 0.05. To detect the association of methylation with gene expression, we manually screened out expression levels of differentially methylated genes.
Functional enrichment analysis of DEGs
To analyse DEGs at the functional level, we performed Gene Ontology (GO) [Citation24] and Kyoto Encyclopedia Gene and Genomes (KEGG pathway) [Citation25] enrichment analyses using the cluster-Profiler package [Citation26]. We identified overrepresented GO terms and KEGG pathways. We chose 0.05 as the threshold for the false discovery rate (FDR) adjusted p-value.
Network analysis
Differently expressed genes contain many cytokine-related genes. Gene network analysis was performed for these cytokine genes. The GenCLiP 3 (http://cismu.net/genclip3/analysis.php/) is an online tool used for literature mining of gene network [Citation27]. GenCLiP 3 was used to construct gene co-occurrence networks of the cytokine genes.
Results
The general pattern of methylation in health controls
Filtered probes that did not meet the requirements according to the default parameters, we detected 767,146 sites in all 44 control samples. The methylation density distribution curve of these methylation sites showed a bimodal distribution (Supplementary Figure S1), where 25.73% were hypomethylated and 49.05% were hypermethylated (β < 0.2 represents hypomethylation, β > 0.8 represents hypermethylation, and β represents the ratio between the signal intensity of the methylated allele and the sum of the methylated and unmethylated alleles).
Among the interrogated sites, 19.02% (145,909/767,146) were located in CpG islands. The mean methylation level for the sites in CpG inlands was significantly lower than that for the sites in the non-CpG inlands (11.25 vs. 57.93).
Differential analysis of peripheral blood cells
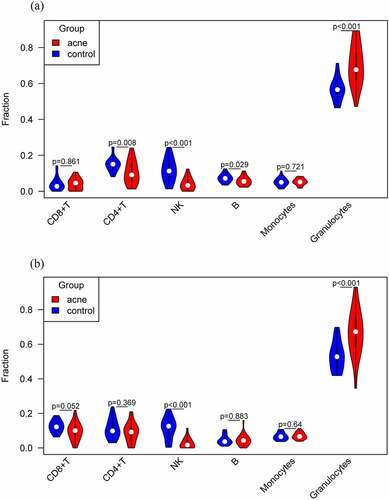
We quantified the cellular composition of each sample (). Differences in six immune cells in blood were compared between patients and healthy controls. Among the 48 samples in stage I, CD4+ T, NK, B, and granulocytes cells were significantly different between the two groups (). In the stage II, NK, and granulocytes cells were significantly different between the two groups (). Taken together, NK, and granulocytes were significantly different between severe acne patients and healthy controls. Among them, the number of NK cells in acne patients is lower than that in healthy people, while the number of granulocytes is the opposite, and the number in acne patients is higher than that in healthy people.
Figure 1. Quantified the cellular composition of peripheral blood cells. (a) Six immune cells of peripheral blood in stage I. (b) Six immune cells of peripheral blood in stage II.
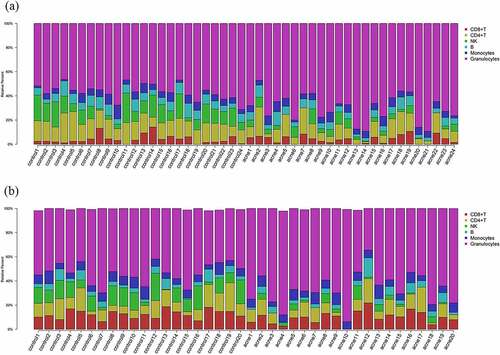
Figure 2. Differences in six immune cells in blood were compared between patients and healthy controls. (a) Differences in stage I. (b) Differences in stage II.
Epigenome-wide association analysis
To identify the loci with differential methylation in severe acne, we carried out an epigenome-wide association analysis between patients and health samples with 24 pairs in stage I. After quality control, 767,494 loci were included in the analysis. PCA was performed using the methylation level of the 767,494 sites. Supplementary Figure S2A showed that acne patients and healthy controls can be largely distinguished. Of course, there was some overlap. It indicated that there were differences in methylation between them, but the differences were not as large as in studies such as cancer samples versus normal samples. After adjustment for cell composition, age, and other factors, the analysis was performed using a linear modelº With a minimum methylation difference of 0.05 between groups and P < 0.05, 504 DMSs were detected in stage I through linear regression model and t-test (Supplementary Table 3). Two DMSs with the most obvious difference (P < 10−7), including cg24836607 (P = 2.60 × 10−7) in the ASZ1 gene and cg15171342 (P = 4.57 × 10−7) located in the TMED3 gene.
We next carried out epigenome-wide association analysis in stage II between another 20 patients and 20 health samples using the same method. 759, 352 loci were included in the analysis after quality control and PCA result was showed in Supplementary Figure S2B. 504 DMSs were identified in stage II (Supplementary Table 4). Two DMSs with the most obvious difference (P < 10−7). The most significant DMS was cg21641816 (P = 5.23 × 10−8), located in the LOC105376375. The second significant DMS was cg20887670 (P = 9.47 × 10−7), which is in the C16orf52 gene.
In total, there were 40 overlapping DMSs in both stages. Significance of overlap was tested using hypergeometric test. The P value (P = 2.96 × 10−69) was less than 0.05, so the overlapping DMSs were not randomly generated and were admissible. Meta-analyses based on inverse variance-weighted fixed-effects were conducted for two stages of the study. The meta-analysis resulted in 23 DMSs (FDR<0.05; and Supplementary Figure S3), of which 16 sites were in 16 genes, and 7 sites were intergenic. Among these 23 DMSs, 52.2% (12 of 23) of DMSs were the hypomethylated and 47.8% (11 of 23) of DMSs were hypermethylated in the acne group. The most significant top three DMSs were cg03020863 located in the PDGFD gene, cg19214707 located in the LOC105375130, and cg24833027 located in the ARHGEF10. The protein encoded by PDGFD plays a critical role in the regulation of cell migration, survival, cell proliferation, and chemotaxis. LOC105375130 belongs to ncRNA gene, and the nearest protein-coding gene is CARD11. It functions as a positive regulator of cell apoptosis and the activation of NF-kappaB. ARHGEF10 codes a Rho guanine nucleotide exchange factor (GEF). Rho GEFs may play a role in capillary barrier function. A sensitivity analysis was performed to determine whether epigenome-wide association results were robust for analysis on M-values instead of β-values. In the QQ plot (), a linear trend is observed, that is, the two results are maximally correlated.
Figure 3. QQ plot to assess whether the epigenome-wide association results were robust to analysis on M-values instead of β-values.
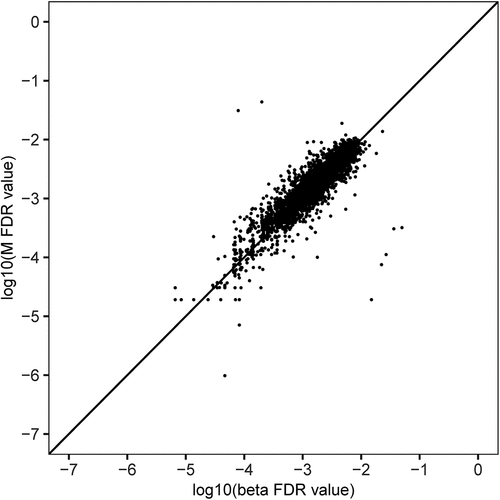
Table 1. Meta-analysis results of differential methylation sites of severe acne in stage I and stage II.
Differentially expressed genes between severe acne and health control
Transcriptome sequencing on five severe acne patients and four health controls was performed. Analysis thresholds set as adjusted P < 0.05 resulted in 1162 differentially expressed genes (DEGs) (Supplementary Table 5 and Supplementary Figure S4). GO and KEGG pathway enrichment analyses were performed for DEGs. The most overrepresented GO terms were showed in Supplementary Figure S5A. They were associated with transcription regulation, and antioxidant activity, among others. According to the KEGG database, genes were mainly enriched for virus infection, among others (Supplementary Figure S5B). Complications and injury to multiple organs caused by virus infection were attributed to be the hyperinflammatory response. A large number of cytokines exert deleterious effects on the body organs. Acne is also a chronic inflammatory disease of the sebaceous follicle. Cytokine, such as IL-8 and TNF-α, releases are also key points in the inflammatory status of human acne lesion. Our results also showed that inflammatory response play an important role in acne.
Differently expressed genes contain many cytokine-related genes. Gene network analysis was performed for these cytokine genes. The co-occurrence network was shown in Supplementary Figure S6. In total, 75 genes were contained in the network. VEGFA, and TP53 were found to be key DEGs in the network, which had a high connectivity degree.
Expression of differentially methylated genes
To identify differentially methylated genes that also differ in gene expression in severe acne, we analysed 16 differentially methylated genes and 1162 differentially expressed genes. We identified a total of 2 DMSs which located in 2 genes, including PARP8, and MAPKAPK2. Among them, MAPKAPK2 was significantly differentially expressed with the largest fold change (log2FC = 1.02), which involved in several cellular processes such as gene expression regulation, cell proliferation, and inflammatory responses.
Discussion
In this study, we performed a genome-wide methylation analysis of 44 pairs of severe acne patients/healthy controls. Since we used a high-density methylation chip, we inspected a great many DMSs by comprehensive analysing of results of stage I and stage II, and finally found that 23 sites were strongly associated with severe acne, of which 10 sites were in protein-coding genes. No association between these 10 genes and acne has been reported before. In gastric cancer, PDGFD regulates the immune microenvironment by modulating the immune genes TAB1 and CXCR4, while lipopolysaccharide (LPS) induced inflammation is mediated by CXCR4, which binds bacterial LPS [Citation28,Citation29]. Severe acne can cause infiltration of inflammatory cells and severe inflammation response under the irritation of Cutibacterium acnes (C. acnes). PDGFD may play an essential role in the regulation of inflammatory response in severe acne. It may activate the NF-κB signalling pathway by regulating the immune gene CXCR4, and eventually lead to the infiltration of inflammatory cells such as neutrophils. ARHGEF10 may play a role in capillary barrier function. A semi-permeable barrier between blood and tissue is maintained by the tight junctions formed among capillary endothelial cells (ECs). As a result of overwhelming inflammation, which occurs in sepsis, these tight junctions (TJs) become disrupted, allowing fluid, proteins, and other molecules to leak into the tissues. The tumour necrosis factor (TNF-α) induces ARHGEF10 to selectively activate RhoB (a small Rho-GTPase), which disrupts the tight junction of microvascular endothelial cells [Citation30]. Mechanistically, the disruption of capillary barrier function may be related to the occurrence of acne. Inflammatory cells need to penetrate from the blood to the skin, which leads to the occurrence of inflammatory cell infiltration in acne. During this process, if the capillary barrier function is impaired, it is more conducive to the migration of inflammatory cells, which exacerbates the inflammatory response of acne. One study also identified ARHGEF10 as a potential biomarker for psoriasis by DNA methylation and gene expression datasets [Citation31]. The IL1R1 gene encodes a receptor for interleukin-1 alpha and beta. It plays a key role in the immune and inflammatory responses induced by cytokines. In chronic, autoinflammatory skin diseases such as psoriasis, the IL-1 signalling pathway plays an important role. The IL-1β-IL-1 R signalling pathway regulates dermal IL-17-producing cells, thereby amplifying inflammatory cascades [Citation32].
We identified a total of 2 differentially methylated genes that also differ in gene expression in acne, including PARP8, and MAPKAPK2. Among them, MAPKAPK2 was significantly differentially expressed with the largest fold change. The MAPK signalling pathway activates MAPKAPK2, a kinase belonging to the MAPKAPK family. P38α activates MAPKAPK2, which controls inflammatory cytokines like TNF-α. MAPKAPK2-mediated activation of ERK1/2 and PI3K signalling is strongly associated with IL-33-induced IL-13 and IL-6 production in psoriasis. Therefore, MAPKAPK2 may be therapeutically useful in suppressing IL-33-induced inflammatory skin diseases such as psoriasis [Citation33]. Importantly, TNF-α serum level was significantly increased in acne patients with severe disease symptoms [Citation34]. The expression of IL-33 was also increased in C. acnes induced cutaneous cell inflammation model [Citation35]. The MAPKAPK2 gene may exacerbate the inflammatory response in acne by controlling the production of the inflammatory factors, such as TNF-α and IL-33. The PARP8 gene has been poorly studied and its association with acne requires further research.
We observed differences in the blood composition between acne patients and healthy controls. Specifically, we observed a reduction in the proportions of NK cell and an increase in the proportions of granulocytes. Granulocytes include neutrophils, eosinophils and basophils, the vast majority of which are neutrophils. Neutrophils mainly target bacteria. When the body is infected with bacteria, neutrophils increase to kill the bacteria. A study showed that Cutibacterium acnes (C. acnes)-induced immune response in murine dermis was regulated by conventional dendritic cells (cDCs) controlling neutrophil recruitment [Citation36]. We can only speculate that our findings reflect non-specific immune response to C. acnes by the acne patients.
Gene network analysis was performed for cytokine genes from DEGs. VEGFA, and TP53 were found to be key DEGs in the network, which had a high connectivity degree. The level of VEGFA was increased during infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), thus promoting inflammation by facilitating recruitment of inflammatory cells [Citation37]. A study identified the gene regulatory network regulated by Vegfa/Nrp1/Flt1 during psoriasis development [Citation38]. Decreased VEGFA was also proven to lower TNF-α, IL-1β, and IL-6 secretion by LPS-induced macrophages [Citation39]. So VEGFA was obtained to be important target for the treatment of acne. A variety of biological processes are regulated by p53 such as cell cycle arrest, apoptosis, senescence and metabolism. Besides these well-known roles of p53, accumulating evidence show that p53 also regulates innate immune and adaptive immune responses. p53 influences the innate immune system by secreted factors that modulate macrophage function to suppress tumourigenesis [Citation40]. The magnitude of p53 expression showed an inverse correlation to acne severity score. Under clinical conditions, isotretinoin treatment significantly increased the expression of p53 in treated patients compared to untreated controls. Isotretinoin induced the expression of p53, which controls multiple transcription factors involved in the pathogenesis of acne vulgaris including FoxO1, androgen receptor and critical genes involved in the induction of autophagy and apoptosis [Citation41].
The meta-analysis resulted in 23 DMSs, of which 7 sites were intergenic. There were also many DMSs located in the intergenic region in stage I and stage II. Intergenic DNA methylation was thought to play an important function in disease. Nongenic methylation was usually associated with transcriptional repression at repetitive elements and transposons. Previous studies suggested that DNA methylation in intergenic regions regulated microRNA expression [Citation42]. Intergenic DMSs can impact transcription via interaction with enhancer elements [Citation43]. Understanding the role of differential methylation in the context of nongenic regions that we did not study, will also be critical. Currently, there are no bioinformatics approaches to identify specific functions for large numbers of intergenic DMSs. Future studies will undoubtedly address many of these important questions concerning the role of DNA methylation in severe acne.
Because of the limited sample size, the statistical efficacy of meta-analysis could not detect more differentially methylated sites. In fact, there were some DMSs worth paying attention to in stage I or stage II. These DMSs are located in cytochrome P450 (CYP), miRNA genes and so on. Although the relationship between these genes and acne vulgaris has not been studied, other CYP family genes have been implicated in acne. A meta-analysis showed that CYP17A1 had potential risk variants for acne presentation and severity across populations [Citation44]. In addition, there were also some studies on the relationship between other genes of CYP family and acne, such as CYP19A1 [Citation45]. Numerous studies have shown that non-coding RNAs play important roles in regulating the pathophysiological processes of acne vulgaris. A study suggested that miR-146a could be a potential player in acne pathogenesis by regulating inflammation, and promoting the lipid production of sebocyte [Citation46]. Several miRNAs play roles in the inflammatory reaction triggered by C. acnes. C. acnes induces miR-146a, which can downregulate the production of IL-6, IL−8, and TNF-α in acne inflammation [Citation47]. A review investigated the current knowledge regarding miRNAs and their link to acne vulgaris. Multiple miRNA (miR-155, miR-223, miR-21, and miRNA-146a) levels were found to be altered in acne vulgaris. These miRNAs are related to pathogenetic aspects, such as the regulation of the innate immune response, regulation of the Th1/Th17 axis, and fibrosis processes that induce scar formation [Citation48].
The limitation of this study is that we do not have more samples to conduct the genome-wide methylation study. Thus, all these findings should be validated in another independent larger sample size population and with pathogenesis investigation. Methylation is also influenced by environmental factors. Although our samples all originated from one province in China, it is true that we cannot completely exclude the influence of environmental factors, such as diet and environmental chemicals. Meanwhile, we should also emphasize that our findings are based on epidemiological hypothesis. These markers can be serviced as a starting point for further studies.
Taken together, we present strong evidence for an association between methylation loci and severe acne in 88 subjects from the Chinese population. From our findings, we show that 23 loci, including PDGFD and ARHGEF10, hold important effects during the onset of severe acne. Furthermore, our data suggest that differentially methylated genes (PARP8 and MAPKAPK2) are also expressed differently in severe acne. Above all, the DMS data generated herein offers precious perspectives on the aetiology of severe acne.
Authors’ contributions
WW, LH, and JY coordinated the overall study and revised the manuscript. HW, TD, and JF prepared DNA for EPIC arrays. HW, and TD analysed the data and drafted the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the medical ethics committee of Dali University (MECDU-201801-12).
Supplemental Material
Download Zip (1.4 MB)Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81760565, and 82160600), Youth Top Talent project of High-level talent development support program of Yunnan Province (YNWR-QNBJ-2020-239), and Li Yunqing expert workstation of Yunnan Province (202005AF150014).
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
Datasets related to this article have been deposited in the OMIX, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn/omix: accession no. OMIX704, and OMIX706).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2199373
Additional information
Funding
References
- Cheng CE, Irwin B, Mauriello D, et al. Self-reported acne severity, treatment, and belief patterns across multiple racial and ethnic groups in adolescent students. Pediatr Dermatol. 2010;27(5):446–11.
- Dreno B. What is new in the pathophysiology of acne, an overview. J Eur Acad Dermatol Venereol. 2017;31(5):8–12.
- Witkowski JA, Parish LC. The assessment of acne: an evaluation of grading and lesion counting in the measurement of acne. Clin Dermatol. 2004;22(5):394–397.
- Desai KP, Martyn-Simmons C, Viner R, et al. Help-seeking behaviours, opportunistic treatment and psychological implications of adolescent acne: cross-sectional studies in schools and hospital outpatient departments in the UK. BMJ Open. 2017;7(9):e016964.
- Al-Shobaili HA, Salem TA, Alzolibani AA, et al. Tumor necrosis factor-alpha -308 G/A and interleukin 10 -1082 A/G gene polymorphisms in patients with acne vulgaris. J Dermatol Sci. 2012;68(1):52–55.
- Ibrahim AA, Salem RM, El-Shimi OS, et al. IL1A (-889) gene polymorphism is associated with the effect of diet as a risk factor in Acne Vulgaris. J Cosmet Dermatol. 2019;18(1):333–336.
- Navarini AA, Simpson MA, Weale M, et al. Genome-wide association study identifies three novel susceptibility loci for severe Acne vulgaris. Nat Commun. 2014;5:4020.
- Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392(10149):777–786.
- Schweiger MR, Kerick M, Timmermann B, et al. The power of NGS technologies to delineate the genome organization in cancer: from mutations to structural variations and epigenetic alterations. Cancer Metastasis Rev. 2011;30(2):199–210.
- Park GT, Han J, Park SG, et al. DNA methylation analysis of CD4+ T cells in patients with psoriasis. Arch Dermatol Res. 2014;306(3):259–268.
- Zhou F, Wang W, Shen C, et al. Epigenome-wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol. 2016;136(4):779–787.
- Jeffries MA, Dozmorov M, Tang Y, et al. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics. 2011;6(5):593–601.
- Mervis JS, McGee JS. DNA methylation and inflammatory skin diseases. Arch Dermatol Res. 2020;312(7):461–466.
- Contassot E, Beer HD, French LE. Interleukin-1, inflammasomes, autoinflammation and the skin. Swiss Med Wkly. 2012;142:w13590.
- Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8(3):389–399.
- Tian Y, Morris TJ, Webster AP, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33(24):3982–3984.
- Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196.
- Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 2012;13:86.
- Balduzzi S, Rucker G, Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid Based Ment Health. 2019;22(4):153–160.
- Ewels P, Magnusson M, Lundin S, et al. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–3048.
- Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915.
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43(Database issue):D1049–1056.
- Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–462.
- Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb). 2021;2(3):100141.
- Wang JH, Zhao LF, Wang HF, et al. GenCLiP 3: mining human genes’ functions and regulatory networks from PubMed based on co-occurrences and natural language processing. Bioinformatics. 2019;36(6):1973–1975.
- Yang Y, Kang W, Yuan Y, et al. Circ-0007707/miR-429/pdgfd pathway regulates the progression of gastric cancer by modulating the immune-gene signature. J Oncol. 2022;2022:2214686.
- Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001;2(4):338–345.
- Khan A, Ni W, Lopez-Giraldez F, et al. Tumor necrosis factor-induced ArhGEF10 selectively activates RhoB contributing to human microvascular endothelial cell tight junction disruption. FASEB J. 2021;35(6):e21627.
- Liu Y, Cui S, Sun J, et al. Identification of potential biomarkers for psoriasis by DNA methylation and gene expression datasets. Front Genet. 2021;12:722803.
- Cai Y, Xue F, Quan C, et al. A critical role of the IL-1beta-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol. 2019;139(1):146–156.
- Drube S, Kraft F, Dudeck J, et al. MK2/3 are pivotal for IL-33-induced and mast cell-dependent leukocyte recruitment and the resulting skin inflammation. J Immunol. 2016;197(9):3662–3668.
- Younis S, Shamim S, Nisar K, et al. Association of TNF-alpha polymorphisms (-857, -863 and -1031), TNF-alpha serum level and lipid profile with acne vulgaris. Saudi J Biol Sci. 2021;28(11):6615–6620.
- Ho HH, Chen CW, Yi TH, et al. Novel application of a Co-Fermented postbiotics of TYCA06/AP-32/CP-9/collagen in the improvement of acne vulgaris-A randomized clinical study of efficacy evaluation. J Cosmet Dermatol. 2022;21(11):6249–6260.
- Janela B, Patel AA, Lau MC, et al. A subset of type i conventional dendritic cells controls cutaneous bacterial infections through VEGFalpha-mediated recruitment of neutrophils. Immunity. 2019;50(4):1069–1083.
- Mosharaf MP, Kibria MK, Hossen MB, et al. Meta-data analysis to explore the hub of the hub-genes that influence SARS-CoV-2 infections highlighting their pathogenetic processes and drugs repurposing. Vaccines (Basel). 2022;10(8):1248.
- Benhadou F, Glitzner E, Brisebarre A, et al. Epidermal autonomous VEGFA/Flt1/Nrp1 functions mediate psoriasis-like disease. Sci Adv. 2020;6(2):eaax5849.
- Lin J, Jiang L, Guo K, et al. Decreased VEGFA alleviates the symptoms of LPS-induced sepsis in a mouse model by inhibiting glycolysis and thereby regulating the polarization of macrophages. Eur J Histochem. 2022;67(1):3528.
- Shi D, Jiang P. A different facet of p53 function: regulation of immunity and inflammation during tumor development. Front Cell Dev Biol. 2021;9:762651.
- Agamia NF, El Mulla KF, Alsayed NM, et al. Isotretinoin treatment upregulates the expression of p53 in the skin and sebaceous glands of patients with acne vulgaris. Arch Dermatol Res. 2022. https://doi.org/10.1007/s00403-022-02508-y
- Luo R, Bai C, Yang L, et al. DNA methylation subpatterns at distinct regulatory regions in human early embryos. Open Biol. 2018;8(10):180131.
- Moody L, Shao J, Chen H, et al. Maternal low-fat diet programs the hepatic epigenome despite exposure to an obesogenic postnatal diet. Nutrients. 2019;11(9):2075.
- Heng AHS, Say YH, Sio YY, et al. Gene variants associated with acne vulgaris presentation and severity: a systematic review and meta-analysis. BMC Med Genomics. 2021;14(1):103.
- Ebrahimi A, Rahimi Z, Ghadami Z, et al. Association between CYP19A<G rs700518 polymorphism with acne vulgaris and its severity: influence on sex hormones level. Int J Mol Cell Med. 2019;8(2):162–168.
- Dull K, Fazekas F, Deak D, et al. MiR-146a modulates TLR1/2 and 4 induced inflammation and links it with proliferation and lipid production via the indirect regulation of GNG7 in human SZ95 sebocytes. Sci Rep. 2021;11(1):21510.
- Zeng R, Xu H, Liu Y, et al. MiR-146a inhibits biofilm-derived cutibacterium acnes-induced inflammatory reactions in human keratinocytes. J Invest Dermatol. 2019;139(12):2488–2496.
- Borgia F, Peterle L, Custurone P, et al. MicroRNA cross-involvement in acne vulgaris and hidradenitis suppurativa: a literature review. Int J Mol Sci. 2022;23(6):3241.