
ABSTRACT
Chronic alcohol consumption may alter mRNA methylation and expression levels of genes related to addiction and reward in the brain, potentially contributing to alcohol tolerance and dependence. Neuron-like (SH-SY5Y) and non-neuronal (SW620) cells were utilized as models to examine chronic intermittent ethanol (CIE) exposure-induced global m6A RNA methylation changes, as well as m6A mRNA methylation changes around the stop codon of three opioid receptor genes (OPRM1, OPRD1, and OPRK1), which are known to regulate pain, reward, and addiction behaviours. CIE exposure for three weeks significantly increased global RNA methylation levels in both SH-SY5Y (t = 3.98, P = 0.007) and SW620 (t = 2.24, P = 0.067) cells. However, a 3-week CIE exposure resulted in hypomethylation around mRNA stop codon regions of OPRM1 and OPRD1 in both cell lines [OPRM1(SH-SY5Y): t = -5.05, P = 0.0005; OPRM1(SW620): t = -3.19, P = 0.013; OPRD1(SH-SY5Y): t = -13.43, P < 0.00001; OPRD1(SW620): t = -4.00, P = 0.003]. Additionally, mRNA expression levels of OPRM1, OPRD1, and OPRK1 were downregulated (corresponding to mRNA hypomethylation) in both SH-SY5Y and SW620 cells after a 3-week CIE exposure. The present study demonstrated that chronic ethanol exposure altered global RNA methylation levels, as well as mRNA methylation and expression levels of opioid receptor genes in both neuron-like and non-neuronal cells. Our findings suggest a potential epitranscriptomic mechanism by which chronic alcohol consumption remodels the expression of reward-related and alcohol responsive genes in the brain, thus increasing the risk of alcohol use disorder development.
Abbreviations: OPRM1: the μ-opioid receptor; OPRD1: the δ-opioid receptor; OPRK1: the κ-opioid receptor; CIE: chronic intermittent ethanol exposure; CIE+WD: chronic intermittent ethanol exposure followed by a 24-hr withdrawal; SH-SY5Y: human neuroblastoma cell Line; SW620: human colon carcinoma cell line; RT-qPCR: reverse transcription followed by quantitative polymerase reaction; MazF-RT-qPCR: MazF digestion followed by RT-qPCR
Introduction
Alcohol use disorder (AUD), which includes alcohol abuse and dependence, is a chronic brain disease characterized by continued alcohol use despite harmful consequences. Family, adoption, and twin studies have demonstrated that genes are responsible for approximately half of the risk of AUD [Citation1]. In addition to genetic variation, environmental factors (such as childhood adversity, stress and anxiety, and social pressure) and chronic alcohol use can also lead to AUD through epigenetic regulation and chromatin remodelling, which cause transcriptional changes in addiction and reward-related genes in specific brain regions and contribute to the endophenotypes (such as alcohol tolerance and dependence) of AUD.
Environmental influences can also lead to epigenetic modifications at post-transcriptional (or RNA) levels. Methylation of N-6 adenosine (m6A) is the most common internal modification of eukaryotic RNAs [Citation2]. m6A sites are enriched near stop codons in 3’UTRs, in long conserved internal exons of mRNAs [Citation3,Citation4], and in the last exons of lncRNAs [Citation3]. RNA m6A modifications can regulate transcript splicing, stability, translation, and microRNA binding. Accumulating evidence suggests that RNA m6A modification can affect the risk of developing various diseases such as brain cancer [Citation5], epilepsy [Citation6], major depressive disorder [Citation7], and stress [Citation8].
Chronic alcohol consumption and AUD can exert adverse effects on the nervous system, resulting in a myriad of symptoms and disorders, including overt neurotoxicity, severe cognitive disruption, depressive episodes, severe anxiety, insomnia, and so on [Citation9]. AUD-associated or alcohol exposure-caused DNA methylation changes and related gene expression changes in peripheral blood or post-mortem brains have been reported [Citation10–12]. However, the research on the impact of AUD or chronic alcohol consumption on RNA methylation is still in its early stages. Our pilot study demonstrated genome-wide RNA methylomic (or epitranscriptomic) changes in the post-mortem nucleus accumbens (NAc) of subjects with AUD [Citation13]. It is hypothesized that alcohol-induced RNA m6A modification regulates the expression of genes associated with reward- and addiction-relevant pathways.
The endogenous opioid system, consisting of three opioid receptors (μ, δ, and κ) and three opioid peptides (β-endorphin, enkephalin, and dynorphin), plays a key role in the rewarding (or addictive) properties of ethanol. The binding of β-endorphin and enkephalin to μ- and δ-opioid receptors on GABAergic neurons removes the tonic inhibition of dopamine release to the nucleus accumbens (NAc) from dopaminergic neurons in the ventral tegmental area (VTA), leading to reward effects. In contrast, the activation of κ-opioid receptors by dynorphin inhibits dopamine release into the NAc, resulting in aversive effects. Acute alcohol consumption stimulates the release of opioid peptides (e.g., β-endorphin) [Citation14]. However, chronic and heavy alcohol consumption can lead to β-endorphin deficiency [Citation15] as well as downregulation of the μ-opioid receptor in the nucleus accumbens and the striatum [Citation16], thus promoting alcohol consumption.
The expression of three opioid receptor genes (OPRM1, OPRD1, and OPRK1) can be influenced by genetic variations and epigenetic changes due to exposure to environmental factors or chronic alcohol consumption. Our previous studies demonstrated significant associations between genetic variants in OPRM1, OPRD1, and OPRK1 and AUD [Citation17,Citation18]. Moreover, we observed a hypermethylated OPRM1 promoter region in subjects with AUD [Citation19] and identified the effect of OPRM1 promoter region DNA methylation on the outcome of naltrexone treatment of AUD [Citation20]. It is likely that chronic alcohol consumption inhibits OPRM1 transcription through hypermethylation of the OPRM1 promoter region. The downregulation of the expression of opioid receptors in AUD subjects may also occur through the mechanism of RNA methylation at the post-transcriptional level. To date, no study has investigated the association between changes in opioid receptor mRNA methylation and AUD. In this study, we used a cellular model to investigate the influence of ethanol exposure/withdrawal on global m6A RNA methylation, as well as m6A modification around mRNA stop codons of OPRM1, OPRD1, and OPRK1. Although the discovery of m6A modifications in mRNA has been long-standing [Citation21], the lack of tools to detect m6A sites has hindered further mRNA methylation research. Recently, a novel endoribonuclease, MazF, was found to be ACA-sequence-specific and sensitive to m6A [Citation22]. When the first A of the ACA sequence in mRNA is methylated, the fragment cannot be cleaved by MazF. As a result, MazF is a powerful tool for investigating m6A modifications in ACA motifs [Citation23,Citation24]. In the present study, we utilized MazF digestion followed by RT-qPCR to quantify methylation levels of m6A sites around the mRNA stop codons of OPRM1, OPRD1, and OPRK1 in neuronal and non-neuronal cells with chronic intermittent ethanol (CIE) exposure and ethanol withdrawal. As m6A modification is enriched near mRNA stop codons [Citation3,Citation4], altered m6A methylation around mRNA stop codons may influence mRNA polyadenylation, stability and degradation, nuclear export, subcellular localization, and translation efficiency. The findings from this research can further our understanding of the novel epitranscriptomic mechanism by which chronic alcohol consumption induces neuroadaptations by altering mRNA methylation levels of addiction- and reward-related genes in brain cells.
Materials and methods
SH-SY5Y and SW620 cells
The human cell lines SH-SY5Y (a neuroblastoma cell line derived from metastatic bone tumour cells) and SW620 (an adenocarcinoma cell line derived from colorectal cancer cells) were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). SH-SY5Y, a neuron-like cell line, has been used in immunology, neuroscience, and toxicology research. SW620, a non-neuronal cancer cell line, has been used in cancer and toxicological research. Both cell types were cultured in a 1:1 mixture of EMEM (ATCC) and F-12K medium (ATCC) with 10% (vol/vol) fetal bovine serum (Corning, Woodland, CA, USA) and 1% penicillin and streptomycin (Corning, Manassas, VA, USA) at 37°C with 5% CO2.
Chronic intermittent ethanol (CIE) exposure and total RNA extraction
A 3-week CIE exposure experiment was conducted as described previously [Citation25] and depicted in Supplementary Figure S1. SH-SY5Y (or SW620) cells (~5 × 105) were seeded in 12 culture dishes (60 × 15 mm) (Corning, Corning, NY, USA). 24 hours after cell plating, six dishes of cells were cultured in medium containing 40 mM ethanol [equivalent to blood ethanol concentration (BEC) after binge drinking [Citation26–28], while another six dishes of cells were cultured in medium without ethanol (i.e., six replicates in both the ethanol treatment group and the control group). After 4 hours, the medium was removed from the 12 dishes (two groups × six replicates per group) and replaced with fresh medium without ethanol. This procedure was repeated three times, from Day 2 (Tuesday) to Day 4 (Thursday). In the following 3 days [Days 5 (Friday), 6 (Saturday), and 7 (Sunday)], the cells in the 12 dishes were cultured in medium without ethanol. This procedure was repeated for the 2nd week [i.e., Day 8 (Monday) – Day 14 (Sunday)] and the first three days of the 3rd week [i.e., Day 15 (Monday) to Day 17 (Wednesday)]. On Day 17, after the six dishes of ethanol-treated cells were exposed to ethanol for 4 h, the cells were harvested and divided into two halves (2 × 6 dishes) and cultured in medium without ethanol. On Day 18 (i.e., Thursday of the 3rd week), six dishes of ethanol-treated cells were exposed to ethanol for 4 hours, and the cells were then harvested (i.e., the group of cells with CIE exposure). Another six dishes of ethanol-treated cells were fed with medium without ethanol for 4 hours, and the cells were then harvested [that is, the group of cells with CIE exposure followed by a 24-hr withdrawal (WD) (CIE exposure + WD)]. Additionally, six dishes of control cells not exposed to ethanol were harvested on Day 18 (i.e., the group of control cells). In total, 18 collections of SH-SY5Y (or SW620) cells (six dishes of CIE-exposed cells, six dishes of CIE-exposed + WD cells, and six dishes of control cells) were obtained and stored at −80°C degree freezer.
Total RNA was extracted from SH-SY5Y (3 × 6 collections) and SW620 cells (3 × 6 collections) using the miRNeasy Mini Kit (QIAGEN, Germantown, MD, USA) and treated with on-column DNase digestion using the RNase-Free DNase Set (QIAGEN, Germantown, MD, USA). The concentration and purity of the extracted RNA samples were examined using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The mean OD260 nm/OD280nm value for the 36 total RNA samples was 2.06 ± 0.03 (mean ± S.D.).
Global m6A RNA methylation assay
The m6A RNA Methylation Quantification Kit (Colorimetric) (ab185912; Abcam, Waltham, MA, USA) was used to detect global m6A RNA methylation levels in the 36 total RNA samples. This is an enzyme-linked immunosorbent assay (ELISA)-like approach. Briefly, 2 μl of extracted total RNAs (diluted to 100 ng/μl) was seeded in each well (containing 80 μl of binding solution) of a clear polystyrene 96-well flat bottom plate (8-well strips on a 12 × 8 frame) and incubated at 37°C for 90 minutes. To prepare a standard curve for m6A quantification, 2 μl of Negative Control and 2 μl of diluted Positive Controls at concentrations of 0.01, 0.02, 0.05, 0.10, 0.20, and 0.50 ng/μl were added to each well (i.e., 0, 0.02, 0.04, 0.1, 0.2, 0.4, and 1 ng of m6A per well) in duplicates. Then, capture antibody solution (50 μl/well), detection antibody solution (50 μl/well), and enhancer solution (50 μl/well) were added sequentially to each well according to the manufacturer’s instructions. After adding 100 μl of developer solution to each well and incubating for approximately 3 minutes at room temperature, the reaction was stopped by adding 100 μl of stop solution to each well. The absorbance at 450 nm was measured within 2–10 minutes using a SpectraMax® i3 Multi-Mode Detection Platform (Molecular Devices, San Jose, CA, USA).
OPRM1, OPRD1, and OPRK1 mRNA stop codon region m6A methylation assay
Approximately 500 ng of total RNA was digested with 20 units of mRNA interferase-MazF enzyme (Takara Bio, Ann Arbor, MI, USA) at 37°C for 30 min in a 10 µL reaction mix containing 1× MazF Buffer and 20 units of RiboLock RNase Inhibitor (Thermo Fisher Scientific, Cambridge, MA, USA). Then, MazF-treated RNAs were reverse transcribed to cDNA using the RevertAid Reverse Transcription Kit (Thermo Fisher Scientific, Cambridge, MA, USA) in a 20 µL reaction mix containing 200 units of RevertAid reverse transcriptase (RT), 20 units of RiboLock RNase Inhibitor, 1 mM of dNTPs, 5 μM of random hexamer primers, and 1× Reaction Buffer. Finally, quantitative polymerase chain reactions (qPCRs) were conducted using the obtained cDNAs as templates to determine the relative m6A methylation levels around the mRNA stop codon regions of the three opioid receptor genes.
Three pairs of primers spanning the mRNA stop codon regions of the three opioid receptor genes (OPRM1, OPRD1, and OPRK1) were designed using NCBI Primer BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/). As shown in Supplementary Figure S2, the OPRM1 primer pair (OPRM1_F:5’ ATTTGAGGCTAAAGAGGGAGGAA 3;’ OPRM1_R:5’ GTCGGAATGGCATGAGACCC 3’) covers a 408-bp sequence that harbours six ACA motifs around the stop codon ‘TAA’ of OPRM1. The OPRD1 primer pair (OPRD1_F:5’ TACGCCAATAGCAGCCTCAA 3;’ OPRD1_R:5’ TGCGCCATTGATCTGTCCTC 3’) covers a 416-bp sequence that harbours two ACA motifs around the stop codon ‘TGA’ of OPRD1. The OPRK1 primer pair (OPRK1_F:5’ CAGAGCACTAGCAGAGTCCG 3;’ OPRK1_R:5’ GAAGAGGTGCATGTGTTGCG 3’) covers a 296-bp sequence that harbours seven ACA motifs around the stop codon ‘TGA’ of OPRK1. Additionally, a pair of primers (ACTB_F:5’ CCCTGGACTTCGAGCAAGAG 3;’ ACTB_R:5’ CCAGGAAGGAAGGCTGGAAG 3’) covering part (144-bp) of the coding sequence of the β-actin gene (ACTB; a housekeeping or reference gene) were designed. Neither the ACTB primer sequences nor the ACTB coding region covered by the ACTB primer pair contained any ACA motifs. The PCR products for the three opioid receptor genes and the reference gene ACTB were visualized by agarose gel electrophoresis (Supplementary Figure S3).
The relative m6A methylation levels around the mRNA stop codon regions of the three opioid receptor genes (OPRM1, OPRD1, and OPRK1) were examined by qPCR using the SYBR® Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA USA) and the QuantStudio™ 12 K Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA USA) with the housekeeping gene ACTB as the reference. The cycling conditions included an initial denaturation stage at 95°C for 15 min, followed by 40 three-step cycles at 94°C for 15 s, then at the optimized annealing temperature (OPRM1: 60°C; OPRD1: 59°C; OPRK1: 51°C) for 30 s, and finally 72°C for 30 s. Melting curve analysis was performed after the qPCRs. The threshold cycles (Ct) of the three opioid receptor genes and ACTB were analysed using the QuantStudio™ 12 K Flex Software.
OPRM1, OPRD1, and OPRK1 mRNA expression assay
The mRNA expression levels of the three opioid receptor genes were determined by RT-qPCR. The experimental procedure was the same as above for measuring the relative mRNA methylation levels of the three opioid receptor genes, with the exception that cDNAs were obtained by direct reverse-transcription of extracted total RNA samples without MazF digestion. The housekeeping gene ACTB was used as a reference gene to determine the relative mRNA expression levels of the three opioid receptor genes.
Statistical analyses
To calculate the global m6A mRNA methylation levels of total RNA samples extracted from SH-SY5Y and SW620 cells, a standard curve was generated by plotting the amount [m6A (ng)] of six diluted Positive Controls at each concentration point versus the OD values [OD450 mm (positive control) – OD450 nm (Negative Control)]. A standard curve and a non-linear regression equation were generated using ELISAcalc software (BioTNT, Shanghai, China) to determine the relationship between the m6A amount (ng) in positive controls and OD450 nm. Among the nonlinear curve models generated by ELISAcalc, the 4-parameter logistic (4PL) curve best modelled the above relationship, and a non-linear regression equation [y = (A – D)/[1 + (x/C)^B] + D; y: m6A amount (ng), x: OD450 nm; A = 0.71469; B = −1.11909; C = 0.03649; D = 0.00040] was obtained (Supplementary Figure S4). The m6A amount (ng) in each total RNA sample was calculated according to this equation using the corresponding OD450 nm value [OD450 nm (RNA samples) – OD450 nm (Negative Control)]. The percentage of m6A in a total RNA sample was calculated using the formula: m6A% = [m6A amount (ng)/S (ng)] × 100% [S: amount (200 ng) of input sample RNA].
As described above, mRNA m6A methylation levels around the mRNA stop codon regions of the three opioid receptor genes were measured by MazF digestion plus RT-qPCR. The relative mRNA methylation levels (ΔCt) of the three opioid receptor genes were normalized to that of the reference gene ACTB [ΔCt = Ct(OPRM1, OPRD1, or OPRK1) – Ct(ACTB)]. Similarly, the relative mRNA expression levels (ΔCt) of opioid receptor genes were normalized to that of the reference gene ACTB [ΔCt = Ct (OPRM1, OPRD1, or OPRK1) – Ct (ACTB)].
An one-way analysis of variance (ANOVA) was used to assess statistically significant differences in the means of RNA methylation/expression levels within and between the three groups of cells (Control, CIE exposure, and CIE exposure followed by a 24-hour ethanol withdrawal). Additionally, post hoc t-tests were performed to compare the means of the RNA methylation/expression levels between each pair of the three groups of cells.
Results
Global m6A RNA methylation changes caused by CIE exposure/withdrawal
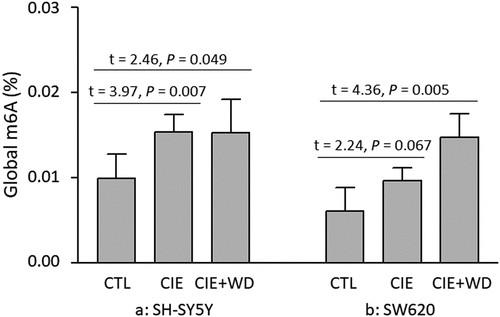
Global m6A RNA methylation changes caused by a 3-week CIE exposure [or a 3-week CIE exposure followed by a 24-hr withdrawal (CIE exposure + WD)] were examined in both neuron-like SH-SY5Y and non-neuronal SW620 cells. The results of the one-way ANOVA suggested a significant impact of ethanol exposure/withdrawal on global m6A RNA methylation levels in both types of cells [SH-SY5Y: F(2,15) = 2.95, P = 0.083; SW620: F(2,15) = 5.12, P = 0.020]. Post hoc pairwise t-tests showed that both CIE exposure and CIE exposure + WD significantly increased the global m6A RNA methylation levels in SH-SY5Y cells (CIE exposure vs. Control: t = 3.98, P = 0.007; CIE exposure + WD vs. Control: t = 2.46, P = 0.049) (). In SW620 cells, CIE exposure also led to elevated global m6A RNA methylation levels (CIE exposure vs. Control: t = 2.24, P = 0.067), and CIE exposure + WD significantly increased the global m6A RNA methylation levels (CIE exposure + WD vs. Control: t = 4.36, P = 0.005) (). In other words, both CIE exposure and CIE exposure + WD resulted in elevated global m6A RNA methylation levels in both neuron-like SH-SY5Y and non-neuronal SW620 cells.
Figure 1. Chronic intermittent ethanol (CIE) exposure/withdrawal-induced global m6A RNA methylation changes. CTL: Control SH-SY5Y or SW620 cells (without ethanol exposure). CIE: a 3-week chronic intermittent ethanol (CIE) exposure. CIE+WD: a 3-week chronic intermittent ethanol exposure followed a 24-hr ethanol withdrawal.
OPRM1 mRNA methylation and expression changes due to CIE exposure/withdrawal
The analysis using an one-way ANOVA revealed a significant effect of ethanol exposure/withdrawal on OPRM1 mRNA methylation [SH-SY5Y: F(2,15) = 46.02, P < 0.00001; SW620: F(2,15) = 5.53, P = 0.018] and expression [SH-SY5Y: F(2,15) = 12.74, P = 0.0006; SW620: F(2,15) = 4.50, P = 0.031] in both types of cells. m6A methylation changes (shown by post hoc t-tests) around the mRNA stop region of OPRM1 due to CIE exposure or CIE exposure + WD are displayed in . Compared to control cells (unexposed to ethanol), CIE exposure led to hypomethylated OPRM1 mRNA in both SH-SY5Y (CIE exposure vs. Control: t = −5.05, P = 0.0005) and SW620 (CIE exposure vs. Control: t = −3.19, P = 0.013) cells. CIE exposure + WD also resulted in OPRM1 mRNA hypomethylation in SW620 cells (CIE exposure + WD vs. Control: t = −2.95, P = 0.016). However, hypermethylated OPRM1 mRNA was observed in SH-SY5Y cells after CIE + WD exposure (CIE exposure + WD vs. Control: t = 5.53, P = 0.0002).
Figure 2. Chronic intermittent ethanol (CIE) exposure/withdrawal-induced mRNA methylation and expression changes in the μ-opioid receptor gene (OPRM1). CTL: Control SH-SY5Y or SW620 cells (without ethanol exposure). CIE: a 3-week chronic intermittent ethanol (CIE) exposure. CIE+WD: a 3-week chronic intermittent ethanol exposure followed a 24-hr ethanol withdrawal.
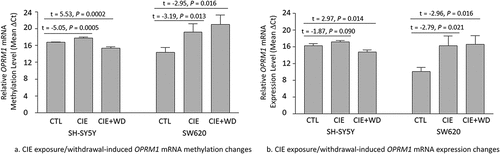
OPRM1 mRNA expression changes (shown by post hoc t-tests) due to CIE exposure or CIE exposure + WD showed a trend like the OPRM1 mRNA methylation changes (). Compared to control cells (unexposed to ethanol), CIE exposure led to downregulation of OPRM1 mRNA expression in both SH-SY5Y (CIE exposure vs. Control: t = −1.87, P = 0.090) and SW620 (CIE exposure vs. Control: t = −2.79, P = 0.021) cells. CIE exposure + WD also resulted in downregulation of OPRM1 mRNA expression in SW620 cells (CIE exposure + WD vs. Control: t = −2.96, P = 0.016). However, OPRM1 mRNA expression was upregulated in SH-SY5Y cells after CIE + WD exposure (CIE exposure + WD vs. Control: t = 2.97, P = 0.014).
OPRD1 mRNA methylation and expression changes due to CIE exposure/withdrawal
The one-way ANOVA showed an significant effect of ethanol exposure/withdrawal on OPRD1 mRNA methylation in both types of cells [SH-SY5Y: F(2,15) = 51.86, P < 0.00001; SW620: F(2,15) = 10.10, P = 0.002] as well as OPRD1 expression in SW620 cells only [SH-SY5Y: F(2,15) = 1.63, P = 0.229; SW620: F(2,15) = 15.86, P = 0.0002]. m6A methylation changes (revealed by post hoc t-tests) around the mRNA stop region of OPRD1 due to CIE exposure or CIE exposure + WD are shown in . Either CIE exposure or CIE exposure + WD led to hypomethylated OPRD1 mRNA in both SH-SY5Y (CIE exposure vs. Control: t = − 13.43, P < 0.00001; CIE exposure + WD vs. Control: t = −7.40, P = 0.00002) and SW620 (CIE exposure vs. Control: t = −4.00, P = 0.003; CIE exposure + WD vs. Control: t = −2.57, P = 0.030) cells.
Figure 3. Chronic intermittent ethanol (CIE) exposure/withdrawal-induced mRNA methylation and expression changes in the δ-opioid receptor gene (OPRD1). CTL: Control SH-SY5Y or SW620 cells (without ethanol exposure). CIE: a 3-week chronic intermittent ethanol (CIE) exposure. CIE+WD: a 3-week chronic intermittent ethanol exposure followed a 24-hr ethanol withdrawal.
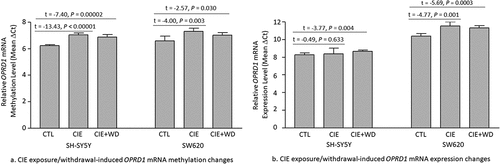
OPRD1 mRNA expression changes (revealed by post hoc t-tests) due to CIE exposure or CIE exposure + WD resembled the changes in OPRD1 mRNA methylation (). Although CIE exposure did not result in significant OPRD1 mRNA expression changes (CIE exposure vs. Control: t = 0.49, P = 0.633) in SH-SY5Y cells, CIE exposure + WD significantly decreased OPRD1 mRNA expression (CIE exposure + WD vs. Control: t = −3.77, P = 0.004) in SH-SY5Y cells. In addition, both CIE exposure and CIE exposure + WD downregulated OPRD1 mRNA expression in SW620 cells (CIE exposure vs. Control: t = − 4.77, P = 0.001; CIE exposure + WD vs. Control: t = − 5.69, P = 0.0003).
OPRK1 mRNA methylation and expression changes due to CIE exposure/withdrawal
The analysis using an one-way ANOVA showed that the impact of ethanol exposure/withdrawal on OPRK1 mRNA methylation [SH-SY5Y: F(2,15) = 1.80, P = 0.199; SW620: F(2,15) = 3.91, P = 0.045] and expression [SH-SY5Y: F(2,15) = 1.44, P = 0.268; SW620: F(2,15) = 4.83, P = 0.027] was significant in SW620 cells only.
Changes in m6A methylation (revealed by post hoc t-tests) around the mRNA stop region of OPRK1 due to CIE exposure or CIE exposure + WD are displayed in . Neither CIE exposure nor CIE exposure + WD caused significant OPRK1 mRNA methylation in SH-SY5Y cells (CIE exposure vs. Control: t = 1.77, P = 0.107; CIE exposure + WD vs. Control: t = 0.66, P = 0.525). Nevertheless, both CIE exposure and CIE exposure + WD led to hypomethylated OPRK1 mRNA in SW620 cells (CIE exposure vs. Control: t = − 2.22, P = 0.054; CIE exposure + WD vs. Control: t = − 2.99, P = 0.015).
Figure 4. Chronic intermittent ethanol (CIE) exposure/withdrawal-induced mRNA methylation and expression changes in the κ-opioid receptor gene (OPRK1). CTL: Control SH-SY5Y or SW620 cells (without ethanol exposure). CIE: a 3-week chronic intermittent ethanol (CIE) exposure. CIE+WD: a 3-week chronic intermittent ethanol exposure followed a 24-hr ethanol withdrawal.
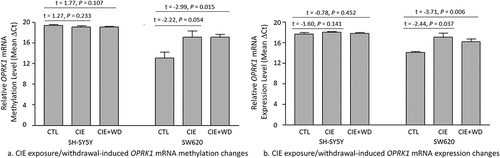
OPRK1 mRNA expression changes (revealed by post hoc t-tests) due to CIE exposure or CIE exposure + WD were similar to the changes in OPRK1 mRNA methylation in both cell lines (). Neither CIE exposure nor CIE exposure + WD caused significant OPRK1 mRNA expression in SH-SY5Y cells (CIE exposure vs. Control: t = −1.60, P = 0.142; CIE exposure + WD vs. Control: t = −0.78, P = 0.452). However, both CIE exposure and CIE exposure + WD significantly downregulated OPRK1 mRNA expression in SW620 cells (CIE exposure vs. Control: t = −2.44, P = 0.037; CIE exposure + WD vs. Control: t = −3.71, P = 0.006).
Discussion
Similar to DNA methylation, the RNA methylation status can also be influenced by external stimuli or environmental factors. RNA methylation changes can fine-tune gene expression at the post-transcriptional (or RNA) level, thus making individuals more adaptable to the environment. In contrast, altered RNA methylation may change transcript fate, leading to pathological behaviours and disorders. There is evidence that RNA m6A modifications can regulate the activity of the dopaminergic brain network [Citation29]. Given the close relationship between the dopamine and opioid systems, we investigated CIE exposure/withdrawal-induced mRNA methylation and expression changes in three opioid receptor genes (OPRM1, OPRD1, and OPRK1) as well as global mRNA methylation changes.
Our study demonstrated that a 3-week CIE exposure or a 3-week CIE exposure followed by a 24-hr withdrawal (CIE exposure + WD) decreased the mRNA methylation levels of three opioid receptor genes in either SY-SY5Y or SW620 cells, although discrepancies in RNA methylation between these two cell lines were observed as well. Briefly, CIE exposure resulted in hypomethylation around the mRNA stop codon regions of OPRM1 and OPRD1 in both cell lines (). Decreased methylation levels of OPRK1 mRNA were also observed in SW620 cells, but not in SH-SY5Y cells after CIE exposure (). Moreover, CIE exposure + WD led to hypomethylation of OPRD1 mRNA in both cell lines and hypomethylation of OPRK1 mRNA in SW620 cells only (but not in SH-SY5Y cells) (). It is likely that the OPRK1 mRNA methylation status in neuron-like SH-SY5Y cells is more resilient to the impact of ethanol exposure/withdrawal than that in non-neuronal SW620 cells. Surprisingly, OPRM1 mRNA was hypermethylated (rather than hypomethylated) in neuron-like SH-SY5Y cells after CIE + WD exposure. The specific reason why CIE + WD causes OPRM1 mRNA hypermethylation is unknown. The mechanism underlying this finding warrants further investigation. In our previous study, we found hypermethylation of OPRM1 promoter DNA sequence in subjects with alcohol use disorder [Citation19]. OPRM1 promoter hypermethylation due to chronic alcohol consumption may inhibit OPRM1 transcription. Presumably, ethanol exposure/withdrawal-induced mRNA methylation changes in opioid receptor genes may regulate opioid receptor gene expression by influencing opioid receptor mRNA metabolism, such as pre-mRNA processing, nuclear export, decay, and translation.
We found that CIE exposure/withdrawal-induced changes in opioid receptor mRNA expression followed the same trend as CIE exposure/withdrawal-induced changes in opioid receptor mRNA methylation in both SH-SY5Y and SW620 cells. Downregulated opioid receptor mRNA expression corresponded to hypomethylated opioid receptor mRNA in both cell lines under both CIE exposure and CIE exposure + WD (). The only exception was that CIE exposure + WD caused upregulation of OPRM1 mRNA expression in neuron-like SH-SY5Y cells, which was consistent with OPRM1 mRNA hypermethylation in SH-SY5Y cells exposed to CIE + WD. The mechanism by which alcohol withdrawal (WD) followed by CIE exposure leads to upregulated (rather than downregulated) OPRM1 mRNA expression is unknown. Among the three opioid receptors, the mu-opioid receptor (OPRM1) is the main target of endogenous opioid peptides and opioid analgesic agents such as beta-endorphin and enkephalins. Previous studies revealed an increase in mu-opioid receptors in the ventral striatum (including the nucleus accumbens) of abstinent AUD subjects, as well as a significant correlation between the severity of alcohol craving and OPRM1 expression levels [Citation30]. These findings are consistent with our observation that alcohol withdrawal induces the upregulation of OPRM1 mRNA expression in neuron-like SH-SY5Y cells.
Although opioid receptor mRNA methylation and expression co-varied in the same direction (i.e., hypomethylation/downregulation or hypermethylation/upregulation) after either CIE exposure or CIE exposure + WD, it remains inconclusive whether altered mRNA methylation directly causes alterations in mRNA expression. In addition to mRNA methylation, other epigenetic modifications (such as DNA methylation, histone modification, and small noncoding RNA regulation) play critical roles in mediating the impact of external stimuli or environmental factors on gene expression. Hence, changes in opioid receptor gene expression associated with CIE exposure or CIE exposure + WD are likely due to the synergistic effect of multiple types of epigenetic modifications. To investigate the biological function of m6A modifications in regulating mRNA expression/translation and cellular processes, advanced epitranscriptomic editing approaches have been developed to edit m6A modifications at targeted sites [Citation31,Citation32]. m6A editing technology can also be applied to pinpoint the function of m6A modifications around the mRNA stop codon regions of opioid receptor genes. However, this type of work is beyond the scope of this study.
This study had three major limitations. First, this study only investigated alcohol exposure/withdrawal-associated m6A methylation changes around the mRNA stop codon regions of the three opioid receptor genes. Although m6A modifications are enriched in 3’ UTRs near stop codons of mRNA, they may occur in other exonic regions of mRNA. For example, m6A modifications in the 5’ UTR can promote cap-independent translation [Citation33]. Therefore, alcohol exposure/withdrawal-induced m6A methylation changes in other exonic regions of opioid receptor mRNA need to be examined. Second, the MazF-qPCR method applied in this study only measured the average methylation level of m6A sites around mRNA stop codon regions rather than the methylation level of a single m6A site, and MazF-qPCR can only determine the methylation level of ACA sites. MazF-qPCR is based on the characteristics of the bacterial ribonuclease MazF, which can cleave single-stranded RNAs immediately upstream of unmodified ACA motifs, but not methylated m6A-CA motifs [Citation22]. In addition to the ACA motif, m6A often exists in exonic regions with a consensus sequence of DRACH (where D = A, G, or U; R = A or G; and H = A, C, or U) [Citation4,Citation34]. To determine the methylation levels of individual m6A sites in the sequences of DRACH, novel approaches, such as miCLIP [Citation35], have been invented to map m6A and the related dimethylated version m6 Am (N6,2’-O-dimethyladenosine) at single-nucleotide resolution in mRNA. Thus, it is necessary to employ high-resolution and high-throughput approaches to profile transcriptome-wide RNA methylation changes induced by CIE exposure or CIE exposure + WD. Third, considering that the RNA methylation status is regulated by m6A writers (adenosine methyltransferases, including METTL3, METTL4, and WTAP), m6A erasers (adenosine demethylases, including FTO and ALKBH5), and m6A readers (such as YTHDF1, YTHDF2, and YTHDF3), our follow-up study needs to explore whether altered opioid receptor mRNA methylation is influenced by ethanol exposure/withdrawal-induced expression changes in the aforementioned m6A regulator genes.
Conclusions
In this study, we used neuron-like and non-neuronal cells as models as well as MazF-RT-qPCR as a tool to analyse global and opioid receptor mRNA stop codon region m6A methylation changes caused by CIE exposure and CIE exposure + WD. We observed increased global RNA m6A methylation levels but mainly decreased mRNA m6A methylation and expression levels of opioid receptor genes in both types of cells with CIE exposure and CIE exposure + WD, although cell type-specific variation was observed as well. The observed increase in global RNA m6A methylation levels due to CIE exposure (or CIE exposure + WD) is consistent with our recent finding that there were more hypermethylated mRNAs than hypomethylated mRNAs in the post-mortem nucleus accumbens of AUD subjects [Citation13]. Given that opioid receptor mRNA methylation and expression co-varied in the same trend, future studies should address the functional role of mRNA methylation in regulating mRNA expression, as well as the cellular processes involved in alcohol-induced neuroadaptations.
Authors’ contributions
YL and HZ conceived and designed the study. Material preparation, data collection, and analysis were performed by YL, JK, and HZ. The first draft of the manuscript was written by HZ. All authors commented on the previous versions of the manuscript. All authors have read and approved the final manuscript.
Supplementary Materials 20231118.docx
Download MS Word (1.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2294515
Data availability statement
The data that support the findings of this study are available upon request from the corresponding author, HZ.
Additional information
Funding
References
- Ducci F, Goldman D. Genetic approaches to addiction: genes and alcohol. Addiction. 2008;103(9):1414–11. doi: 10.1111/j.1360-0443.2008.02203.x
- Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci, USA. 1974;71:3971–5. doi: 10.1073/pnas.71.10.3971
- Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46. doi: 10.1016/j.cell.2012.05.003
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112
- Cui Q, Shi H, Ye P, et al. m(6)A RNA methylation regulates the Self-Renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34. doi: 10.1016/j.celrep.2017.02.059
- Rowles J, Wong M, Powers R, et al. FTO , RNA epigenetics and epilepsy. Epigenetics. 2012;7(10):1094–1097. doi: 10.4161/epi.21977
- Du T, Rao S, Wu L, et al. An association study of the m6A genes with major depressive disorder in Chinese han population. J Affect Disord. 2015;183:279–286. doi: 10.1016/j.jad.2015.05.025
- Engel M, Eggert C, Kaplick PM, et al. The role of m(6)A/m-RNA methylation in stress Response regulation. Neuron. 2018;99:389–403 e9. doi: 10.1016/j.neuron.2018.07.009
- Schuckit MA. Alcohol-use disorders. Lancet. 2009;373(9662):492–501. doi: 10.1016/S0140-6736(09)60009-X
- Mahna D, Puri S, Sharma S. DNA methylation signatures: biomarkers of drug and alcohol abuse. Mutat Res Rev Mutat Res. 2018;777:19–28. doi: 10.1016/j.mrrev.2018.06.002
- Zhang H, Gelernter J. Review: DNA methylation and alcohol use disorders: progress and challenges. Am J Addict. 2017;26:502–15. doi: 10.1111/ajad.12465
- Gatta E, Grayson DR, Auta J, et al. Genome-wide methylation in alcohol use disorder subjects: implications for an epigenetic regulation of the cortico-limbic glucocorticoid receptors (NR3C1). Mol Psychiatry. 2021;26:1029–41. doi: 10.1038/s41380-019-0449-6
- Liu Y, Zhang H. RNA m6A modification changes in postmortem nucleus accumbens of subjects with alcohol use disorder: a Pilot study. Genes (Basel). 2022;13(6):958. doi: 10.3390/genes13060958
- Herz A. Endogenous opioid systems and alcohol addiction. Psychopharmacol (Berl). 1997;129(2):99–111. doi: 10.1007/s002130050169
- Gianoulakis C. Influence of the endogenous opioid system on high alcohol consumption and genetic predisposition to alcoholism. J Psychiatry Neurosci. 2001;26:304–18.
- Turchan J, Przewlocka B, Toth G, et al. The effect of repeated administration of morphine, cocaine and ethanol on mu and delta opioid receptor density in the nucleus accumbens and striatum of the rat. Neuroscience. 1999;91(3):971–977. doi: 10.1016/S0306-4522(98)00637-X
- Zhang H, Luo X, Kranzler HR, et al. Association between two mu-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum Mol Genet. 2006;15:807–19. doi: 10.1093/hmg/ddl024
- Zhang H, Kranzler HR, Yang BZ, et al. The OPRD1 and OPRK1 loci in alcohol or drug dependence: OPRD1 variation modulates substance dependence risk. Mol Psychiatry. 2008;13(5):531–543. doi: 10.1038/sj.mp.4002035
- Zhang H, Herman AI, Kranzler HR, et al. Hypermethylation of OPRM1 promoter region in European Americans with alcohol dependence. J Hum Genet. 2012;57(10):670–675. doi: 10.1038/jhg.2012.98
- Lin Y, Kranzler HR, Farrer LA, et al. An analysis of the effect of mu-opioid receptor gene (OPRM1) promoter region DNA methylation on the response of naltrexone treatment of alcohol dependence. Pharmacogenomics J. 2020;20(5):672–680. doi: 10.1038/s41397-020-0158-1
- Niu Y, Zhao X, Wu YS, et al. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Int J Genomics Proteomics. 2013;11(1):8–17. doi: 10.1016/j.gpb.2012.12.002
- Imanishi M, Tsuji S, Suda A, et al. Detection of N(6)-methyladenosine based on the methyl-sensitivity of MazF RNA endonuclease. Chem Commun (Camb). 2017;53:12930–3. doi: 10.1039/C7CC07699A
- Pandey RR, Pillai RS. Counting the cuts: MAZTER-Seq quantifies m(6)A levels using a methylation-sensitive ribonuclease. Cell. 2019;178:515–7. doi: 10.1016/j.cell.2019.07.006
- Chen HX, Zhang Z, Ma DZ, et al. Mapping single-nucleotide m(6)A by m(6)A-REF-seq. Methods. 2022;203:392–8. doi: 10.1016/j.ymeth.2021.06.013
- McClintick JN, Thapa K, Liu Y, et al. Effects of chronic intermittent ethanol exposure and withdrawal on neuroblastoma cell transcriptome. Alcohol. 2020;85:119–126. doi: 10.1016/j.alcohol.2019.12.004
- Bell RL, Rodd ZA, Engleman EA, et al. Scheduled access alcohol drinking by alcohol-preferring (P) and high-alcohol-drinking (HAD) rats: modeling adolescent and adult binge-like drinking. Alcohol. 2014;48(3):225–234. doi: 10.1016/j.alcohol.2013.10.004
- Jury NJ, Pollack GA, Ward MJ, et al. Chronic ethanol during adolescence impacts corticolimbic dendritic spines and behavior. Alcohol Clin Exp Res. 2017;41(7):1298–1308. doi: 10.1111/acer.13422
- Melendez RI. Intermittent (every-other-day) drinking induces rapid escalation of ethanol intake and preference in adolescent and adult C57BL/6J mice. Alcohol Clin Exp Res. 2011;35(4):652–658. doi: 10.1111/j.1530-0277.2010.01383.x
- Hess ME, Hess S, Meyer KD, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–8. doi: 10.1038/nn.3449
- Heinz A, Reimold M, Wrase J, et al. Correlation of stable elevations in striatal mu-opioid receptor availability in detoxified alcoholic patients with alcohol craving: a positron emission tomography study using carbon 11-labeled carfentanil. Arch Gen Psychiatry. 2005;62:57–64. doi: 10.1001/archpsyc.62.1.57
- Xia Z, Tang M, Ma J, et al. Epitranscriptomic editing of the RNA N6-methyladenosine modification by dCasrx conjugated methyltransferase and demethylase. Nucleic Acids Res. 2021;49(13):7361–7374. doi: 10.1093/nar/gkab517
- Liu XM, Zhou J, Mao Y, et al. Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol. 2019;15:865–71. doi: 10.1038/s41589-019-0327-1
- Meyer KD, Patil DP, Zhou J, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012
- Kane SE, Beemon K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol. 1985;5(9):2298–2306. doi: 10.1128/MCB.5.9.2298
- Linder B, Grozhik AV, Olarerin-George AO, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–772. doi: 10.1038/nmeth.3453