
ABSTRACT
Accurately identifying life-threatening prostate cancer (PCa) at time of diagnosis remains an unsolved problem. We evaluated whether DNA methylation status of selected candidate genes can predict the risk of metastasis beyond clinical risk factors in men with untreated PCa. A nested case-control study was conducted among men diagnosed with localized PCa at Kaiser Permanente California between 01/01/1997–12/31/2006 who did not receive curative treatments. Cases were those who developed metastasis within 10 years from diagnosis. Controls were selected using density sampling. Ninety-eight candidate genes were selected from functional categories of cell cycle control, metastasis/tumour suppressors, cell signalling, cell adhesion/motility/invasion, angiogenesis, and immune function, and 41 from pluripotency genes. Cancer DNA from diagnostic biopsy blocks were extracted and analysed. Associations of methylation status were assessed using CpG site level and principal components-based analysis in conditional logistic regressions. In 215 cases and 404 controls, 27 candidate genes were found to be statistically significant in at least one of the two analytical approaches. The agreement between the methods was 25.9% (7 candidate genes, including 2 pluripotency markers). The DNA methylation status of several candidate genes was significantly associated with risk of metastasis in untreated localized PCa patients. These findings may inform future risk prediction models for PCa metastasis beyond clinical characteristics.
KEYWORDS:
Introduction
Prostate cancer (PCa) is the most common cancer in men in the United States [Citation1]. Approximately 80% of PCa cases are diagnosed at the localized stage, of which many are indolent and will not benefit from curative treatment [Citation2,Citation3]. Current clinical guidelines recommend active surveillance, rather than curative treatment, as the preferred care option for localized PCa patients with a “very low-risk profile”. Active surveillance can also be considered as a treatment option for other localized PCa[Citation4]. However, a small proportion of localized PCa can progress to metastasis[Citation5]. As such, there remains a need to identify PCa cases whose metastasis may be prevented by curative treatments at the localized stage. To this end, biomarkers that are closely related to tumour biology may help with the development of risk stratification algorithms for localized PCa and may also help inform the development of novel therapeutic targets.
Epigenetic regulation is a major mechanism underlying tumour heterogeneity and disease progression [Citation6,Citation7]. Of all epigenetic regulation mechanisms, DNA methylation is the most stable and readily measurable for use as a clinical epigenetic marker [Citation8,Citation9]. Recent studies have demonstrated that aberrant DNA methylation of numerous genes, including metastasis suppressors and genes that maintain cell differentiation status, is a major mechanism underlying tumour metastatic progression [Citation6,Citation7,Citation10]. Aberrant methylation of the promoter regions of tumour suppressor genes has been found in many cancer types that suppress key functions that prevent cancer occurrence, such as cell cycle control, metastasis and tumour suppression, cell signalling and transcription, cell adhesion, motility, invasion, angiogenesis, and immune function and inflammation [Citation10,Citation11].
In addition to the genes in the important functional categories thought to promote metastasis, the cancer stem cell (CSC) theory has emerged as an appealing alternative model for cancer metastasis that may also be influenced by aberrant DNA methylation [Citation12,Citation13]. CSCs refer to a subset of tumour cells that share features with normal stem cells in pluripotency and the ability to self-renew [Citation14,Citation15]. The CSC hypothesis states that this subset of cancer cells within each tumour is long-lived and responsible for tumour growth, maintenance, and metastasis. Evidence supporting the role of CSC in metastasis is accumulating. For example, studies have reported that PCa cell lines depleted of CSC-like cells were abrogated in their metastatic potential [Citation16,Citation17] and gene expression signatures in aggressive tumours, including metastatic PCa cells, are similar to those of embryonic stem cells [Citation18] To date, the literature suggests that higher expression of stem cell markers is associated with worse clinical outcomes [Citation16–20]. We thus hypothesize that DNA methylation profiles of stem cell identity, such as hypo-methylation of pluripotency genes, are predictive of PCa metastasis. However, the role of stem cell markers in PCa progression has not been widely studied. Thus, measuring the methylation of these genes may allow for the opportunity to improve PCa prognosis [Citation21–23].
Recently, a fair number of studies have searched for DNA methylation markers predictive of PCa disease progression[Citation5]. Most of which used tissue from PCa patients who have been treated with radical prostatectomy and evaluated biochemical recurrence as their primary outcome of interest [Citation7,Citation8]. As such, methylation markers measurable at the time of diagnosis that predict the risk of metastasis in the absence of treatment remain to be determined. Thus, studies of prognostic methylation markers that focus on the natural disease course among untreated men are critically needed to inform treatment decisions for those with localized PCa.
To address this gap in the literature, we conducted a nested case-control study to evaluate the association between DNA methylation markers measured using diagnostic biopsy tissues and the risk of metastasis in men diagnosed with localized PCa who did not receive curative treatment. We hypothesized that tumour DNA methylation of specific candidate genes is predictive of PCa metastatic progression beyond routine clinical and pathological prognostic factors in men with localized PCa. Here we used a candidate gene approach instead of a genome-wide scan as true biological signals do not always result in the largest effect sizes, making them difficult to find in agnostics scans. We have previously conducted a genome-wide scan with findings (non-overlapping) reported separately[Citation24].
Methods
Study setting, design, and population
This study was conducted at Kaiser Permanente Southern California (KPSC) and Kaiser Permanente Northern California (KPNC). KPSC and KPNC are integrated healthcare delivery systems that provide comprehensive health services for over nine million members. KP members are racially and socioeconomically diverse, and broadly representative of California residents. The high KP membership retention rate permitted long-term follow-up of metastasis outcomes in the study; 88% of PCa patients who remained alive 10 years after PCa diagnosis retained their KP membership.
A nested case-control study was conducted to evaluate the association between DNA methylation markers and the risk of metastasis among men diagnosed with localized PCa who did not receive curative treatment. The source cohort was identified using the following inclusion criteria: (1) men diagnosed with PCa at KPSC or KPNC between 1 January 1997 and 31 December 2006, and (2) diagnosed at the localized stage (stage I). Men who met the following criteria were excluded: (1) unknown Gleason score at diagnosis; (2) received active treatment for PCa, including prostatectomy, radiation therapy, hormone therapy, or chemotherapy, within six months of diagnosis; and (3) identified to have metastatic disease, died, or were lost to follow-up within six months of diagnosis. All eligible men were followed up from 6 months after PCa diagnosis to identify metastasis and censored at the initiation of PCa treatment (prostatectomy, radiation therapy, chemotherapy, or immunotherapy), non-PCa-related death, KPSC membership termination, or end of follow-up period (10 years after initial PCa diagnosis), whichever came first.
Cases included all men with PCa metastasis identified during the study follow-up period. Controls were selected from the source cohort using the density sampling method and individually matched to cases at up to a 3:1 ratio by age (within 5 years), race (black vs. non-black), year of PCa diagnosis (within 5 years), and KP region (KPSC or KPNC). Additional controls were added to ensure that each case had at least two controls matched according to the Gleason score. If the selected controls did not have tissue blocks available, we selected additional controls to ensure that each case had at least two controls with a tumour block, if possible, while maintaining the matching criteria.
Data collection
Men diagnosed with PCa were identified from KPSC’s and KPNC’s Surveillance, Epidemiology and End Result (SEER) affiliated cancer registries. The KPSC and KPNC cancer registries collect data on the clinical stage, Gleason grade, year of diagnosis, age at diagnosis, race/ethnicity, and initial course of cancer treatment.
Potential metastases were identified using an algorithm based on ICD-9 and ICD-10 diagnosis codes for metastatic malignancy and disseminated cancer; natural language processing of radiology reports (including bone scans); serum prostate-specific antigen (PSA) levels >20 ng/ml; CPT procedure codes for treatment initiation and utilization of chemotherapy, immunotherapy, or hormonal therapy (leuprolide); encounters with an oncologist 12 months after diagnosis; and death due to PCa. Manual chart reviews were conducted to identify probable cases identified by the algorithm to confirm PCa metastasis and date of metastasis diagnosis. A random sample of 10% of the charts was reviewed a second time to ensure the quality of the chart review. An experienced urologist reviewed all questionable cases and made a final determination on the presence of metastasis.
Initiation of curative treatment, prostatectomy, brachytherapy, external beam radiation therapy, chemotherapy, and immunotherapy were identified using ICD-9, ICD-10, and CPT procedure codes in the electronic health records, as well as from the KPSC and KPNC cancer registries. Data on PSA levels at diagnosis were obtained from KP’s laboratory database. Deaths were identified from clinical records and death certificate linkage with State of California and Social Security Administration records. PCa mortality was identified as PCa being the underlying cause of death on the death certificate.
DNA methylation markers
A literature review was conducted and 100 candidate genes linked to prostate and/or breast cancer metastasis were selected to be simultaneously evaluated in this study (). These genes were selected from the functional categories of (1) cell cycle control (2) metastasis/tumour suppressors (3) cell signalling and transcription factors (4) cell adhesion, motility, or invasion (5) angiogenesis and (6) immune function/inflammation. Genes that were not found in our array were omitted from the data analysis (APC, HSP90, and eNOS), resulting in 98 candidate genes being included in the analysis.
Table 1. Candidate genes selected for prognostic evaluation and results.
In addition, we selected 44 stem cell markers from two sets of genes: (1) pluripotency genes [Citation19] and (2) genes overexpressed in human embryonic stem cells, as reported in a meta-analysis of 8 profiling studies [Citation75]. Stem cell marker genes that were not found in our array were omitted from the data analysis (OCT4, SOX2, and POUSF1), resulting in the inclusion of 41 stem cell genes in the analysis.
Pathology review
Archived diagnostic biopsy H&E slides and formalin-fixed, paraffin-embedded (FFPE) blocks for study subjects were retried from KP’s centralized storage centre. Pathologists centrally reviewed all pathology reports and diagnostic slides to identify appropriate biopsy cores for the methylation assay. The cancerous area is circled directly on each FFPE block for macrodissection. Microdissection was not performed for the following reasons: (1) gene expression studies suggest an important role of stromal cells in the metastasis signature; (2) micro-dissection is limited for routine clinical use due to costs; and (3) PCa tumour glands are usually clustered in biopsy cores, making macro-dissection easy to perform and contamination less of a concern. When necessary, multiple biopsy cores were used to obtain sufficient DNA samples for the methylation assays.
Laboratory procedures
We used Illumina’s Infinium Methylation EPIC BeadChip following the manufacturer’s protocol for genome-wide methylation profiling. DNA extraction was performed using the Qiagen QIAamp DNA FFPE Tissue Kit. Purified DNA was quantified using the PicoGreen DNA quantification assay, stored at −20°C, and shipped via overnight shipping with dry ice packaging to the University of Southern California Core Facility for the methylation assay. At the Core Facility, purified DNA was bisulphite-converted (Zymo’s EZ DNA methylation kit), treated with a FFPE DNA Restoration Kit, and evaluated using Illumina’s FFPE QC Kit. The methylation status of the interrogated CpG site was presented as the β value, which is a continuous variable ranging from 0 (unmethylated) to 1 (fully methylated). To avoid confounding due to batch, BeadChip, or well position effects, we ensured that each case and his matched controls were included in the same batch and BeadChip. Samples within the case and matched controls were randomly assigned to well positions. Background correction and dye-bias normalization were performed at the USC core lab using the ‘noob’ function in the minfi R package. The ‘noob’ function corrects for background fluorescence intensities and red-green dye-bias[Citation76].. The MethylationEPIC array targeted a total of 5273 CpGs from the 139 candidate genes.
Statistical analysis
Data points with a probe failure rate of > 5% were omitted from the analysis. Because one of the main purposes of this study was to evaluate the added predictive value of methylation markers, methylation signals that may be due to SNPs were not filtered.
The distributions of the demographic and clinical characteristics, such as age at diagnosis, race, year of diagnosis, Gleason grade, serum PSA level, number of positive biopsy cores, and chronic comorbid conditions, were first examined in cases and controls using t-tests or chi-square tests. The distribution of the time from PCa diagnosis to diagnosis of metastatic disease among cases and the cumulative incidence of metastasis in this untreated cohort were also calculated (note that the cumulative incidence of metastasis in the source cohort has been described in prior publication)[Citation3].
Epigenetic associations were assessed using two statistical methods: CpG site-level and principal components (PC)-based analyses. Conditional logistic regression was used to test the associations between methylation levels and metastasis status, adjusting for potential confounders (age at diagnosis, race, year of diagnosis, Gleason score, serum PSA level, number of positive biopsy cores, and chronic comorbid conditions). Despite age and race being matching factors, they were included as covariates to account for potential residual confounding effects in the analysis.
In the CpG site-level analysis, each CpG site in the candidate genes was tested individually. To control for multiple comparisons, given that the Bonferroni correction is known to be highly conservative with reduced power, especially when the tests are related, we applied a double false discovery rate (DFDR) procedure [Citation77] to adjust the p-values. The DFDR approach (named `new DFDR’) [Citation77] controls the expected frequency of incorrectly rejected null hypotheses at the gene level. We used the Benjamini and Hochberg (BH) procedure and a threshold of 0.1 to control the false-discovery rate at 10%. In the DFDR procedure, BH adjustment was applied to the site-level p-values for each gene region, and the minimum BH-adjusted p-value was recorded. These minimum BH-adjusted p-values underwent a subsequent BH adjustment, and gene regions with a twice-corrected p-value less than 0.1 are selected as significant genes. Finally, the unadjusted p-values for the CpG sites from these selected genes were subjected to a single FDR adjustment. Sites with adjusted p-values less than 0.10 were considered significant sites in the analysis.
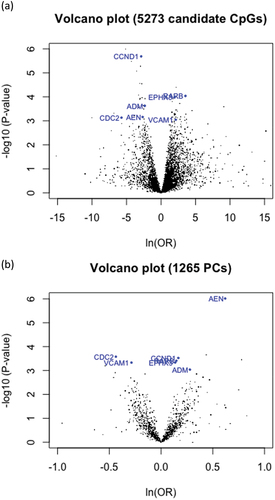
In PC-based analysis, gene-level PCs were computed using all CpG variants within a gene. Gene-level PCs were tested using a conditional logistic model to explore epigenetic associations with metastasis status after adjusting for covariates. For each gene, the top PCs were selected so that the sum of their variances explained 80% of the total variance. If the 80% threshold cannot be satisfied by the top ten principal components, suggesting the sites shared low correlation, then the top ten PCs were selected to achieve a balance between maximizing the variance and minimizing the number of tests. PC-level significance was determined by testing the regression coefficients of PCs using Wald tests. Similar to the CpG site-level analysis, DFDR was applied using a false-discovery level of 0.1 to select the significant genes and their significant PCs. Volcano plots were created to display the univariate results of all individual CpG and gene-level PC tests ().
For the candidate genes that were identified by both the CpG-site level analysis and the PC-based methods, we created figures of distributions of beta-values across CpG sites assayed for each of these genes by case and control status (Supplemental Figure S1) to allow a visual inspection for differentially methylated regions. Cubic spline of average beta-values was created separately for cases and controls and smoothed across the entire gene[Citation78]. We also created heatmaps for these same candidate genes for cases and controls to offer an alternative approach for data visualization (Supplemental Figure S2).
To assess whether the candidate genes found to be significantly associated with metastasis (by both methods) improved prediction of metastasis development beyond established clinical risk factors, we calculated the c-statistics, or the area under the receiver operating characteristic curve (AUC), for the model containing clinical risk factors alone, and for the model containing both clinical risk factors and the methylation status of candidate genes (using the PCs). The clinical risk factors were modelled as a single a single clinical risk score composed of age at diagnosis, race/ethnicity, stage at diagnosis, Gleason score, PSA at diagnosis, PSA doubling time, and number of positive biopsy cores. The AUCs from these two models were compared using likelihood ratio test. The receiver operating characteristics (ROC) curves were generated for prediction models containing: methylation status of each of these candidate genes, methylation status of all these candidate genes, the clinical risk score alone, and the clinical risk score + methylation status of the candidate genes. Given the nested case-control study design, all AUC and ROC curves were generated using a weighted approach, with the weight being 1 for cases and the inverse of the control sampling probability for controls. Data cleaning was conducted using SAS Enterprise Guide version 7.1 statistical software and the analyses were conducted using the R programming language.
Results
A total of 45,121 men diagnosed with PCa between 1997–2006 were identified at KPSC and KPNC, of whom 39,802 (88.2%) were diagnosed at localized stages. After the exclusion criteria, the source cohort for the case-control selection consisted of 10,039 men (). Of these men, 357 metastatic cases were confirmed by chart review, and 1,322 matched controls were selected. Among the identified cases and controls, 1,185 had at least one tissue block retrieved for the diagnostic biopsy, of which 710 had sufficient DNA samples extracted, including 232 cases and 478 controls. A total of 215 cases and 404 controls yielded successful methylation results.
Figure 1. Study population flow chart.
The mean age at PCa diagnosis was 72.7 years for cases and 72.9 years for controls (). Among the cases, approximately 97% were diagnosed at stage I and 3.2% were diagnosed with stage II cancer, whereas among controls 98.8% were diagnosed at stage I and 1.2% at stage II. Approximately 42% of the cases had a Gleason score of 7, and 26% had a Gleason score of ≤6. In contrast, approximately 27% and 51% of controls had Gleason scores of 7 and ≤6, respectively. Cases also had significantly higher PSA levels at diagnosis, shorter PSA doubling time, and a greater number of biopsy cores positive for cancer than controls (). In the overall cohort, the cumulative incidence of metastasis was 7.8% over 10 years. The total mean follow-up times were 6.8 years for cases (median = 7.2 years, Q1-Q3 = 4.6–9.8 years) and 9.6 years for controls (median = 10.0 years, Q1-Q3 = 7.3–10.0 years).
Table 2. Demographic and clinical characteristics of the study population.
The genes selected for prognostic evaluation are shown in . The individual CpG site-level results are displayed in . After DFDR correction, CpG sites in the following 24 candidate genes were found to be significant: ADM, AEN, CCND1, CDC2, CPN1, DNMT3A, EPHA2, EPHX3, FBLN2, FGFR2, GINS4, ICAM1, L1TD1, MATN2, ORC2L, PRDM14, PTN, RAP1GAP, RARB, RASSF1A, TGFB1, TMSB15B, VCAM1, and WSCD1 ( and Supplemental Table S1). For all significant individual CpG sites in the candidate genes ADM, AEN, CCND1, CDC2, ORC2L, RAP1GAP, RASSF1A, and TGFB1, higher methylation was associated with a lower risk of metastasis. For all significant individual CpG sites in CPN1 and EPHX3 higher methylation was associated with a higher risk of metastasis. Higher methylation in ICAM1, RARB, and VCAM1 were associated with a higher risk of metastatic cancer in the most statistically significant individual CpG sites, but were associated with a lower risk of metastatic cancer in some other individual CpG sites.
Figure 2. Volcano plots for the (a) CpG site-level analysis and (b) PC-based analysis.
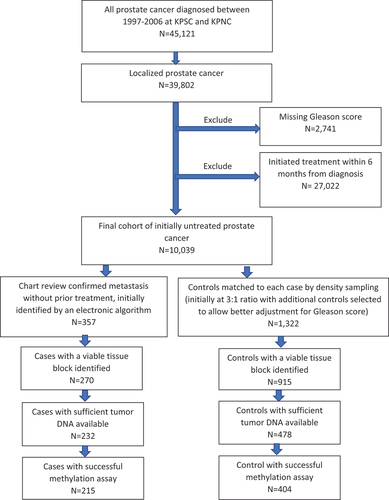
Table 3. Twenty-four candidate genes found to be significant in the CpG site-level analysis.
In the PC-based analysis, PCs in ten candidate genes were found to be significant: ADM, AEN, CCND1, CDC2, CTNNB1, DDAH1, EPHX3, MYBL2, RARB, and VCAM1 (, Supplemental Table S1, and Figure S2b). The principal components of the candidate genes, AEN, CDC2, and VCAM1, were negatively associated with metastatic cancer (OR < 1.0). The principal components of the candidate genes, ADM, CCND1, CTNNB1, EPHX3, and RARB, were positively associated with metastatic cancer (OR > 1.0).
Table 4. Candidate genes found to be significant in PC-based analysis.
The two methods combined identified 27 of the 139 candidate genes to be significant. The agreement between the CpG site-level analysis and PC-based analysis was 7 candidate genes: ADM, AEN, CCND1, CDC2, EPHX3, RARB, and VCAM1 (). That is, the agreement between both methods was 7 of the 27 total significant candidate genes (25.9%). The seven candidate genes identified in our study fell into the following functional categories: cell cycle control (CCND1 and CDC2), metastasis suppressors and tumour suppressors (RARB), cell adhesion/motility/invasion (VCAM1), angiogenesis (ADM), and genes overexpressed in human embryonic stem cells (AEN and EPHX3). No candidate genes were found to be statistically significant in the following functional categories: cell signalling and transcription factors, immune function/inflammation, and pluripotency genes (see Supplemental Table S1 for a full list of the results).
The evaluation of the graphs depicting beta-value distributions (Supplemental Figure S1) and the heatmaps (Supplemental Figure S2) did not suggest largely distinctive patterns between cases and controls, with considerable variability in CpG methylation within each group. Inclusion of the PCs of the 7 candidate genes identified by both the CpG site-level analysis and PC-based analysis into the risk prediction model increased the AUC from 0.70 to 0.74 (p = 0.001) (clinical risk score only and clinical risk score+ methylation status of the 7 candidate genes). The ROC curves for these two risk prediction models are shown in Supplemental Figure S3, along with the ROC curves for each individual gene, and for all 7 genes.
Discussion
In this study, we examined 139 candidate genes to identify tumour DNA methylation sites that can help predict metastatic progression beyond routine clinical and pathological prognostic factors in men with localized, untreated PCa. We identified 27 candidate genes from CpG sites and PC-based analyses to be significantly associated with risk of metastasis among men with localized PCa who did not receive curative treatment. Seven of these candidate genes overlapped significantly between the two methods used. Adding methylation status of these seven candidate genes in the form of PCs to the logistic regression model significantly improved the model discrimination ability of cases and controls, as suggested by the improved AUC statistics comparing the model with both the 7 candidate genes and clinical risk factors, and the model with clinical risk factors alone. Our findings suggest that these seven candidate genes and their corresponding significant methylation sites may help identify localized PCa patients who are at a higher risk of metastasis beyond clinical characteristics.
Of the seven genes identified, the cell cycle control genes CCND1 and CDC2 are related to cell cycle regulation. Overexpression of these genes are known to prevent normal cell cycle regulation, causing uncontrolled cell proliferation and abnormal tissue growth [Citation16,Citation26,Citation27]. The metastasis suppressor and tumour suppressor gene identified (RARB) is important for regulating cell proliferation, differentiation, migration, and apoptosis[Citation39]. Underexpression of this gene may lead to tumour growth and metastasis. The cell adhesion gene identified (VCAM1) is related to signalling pathways that facilitate tumour progression and metastasis[Citation61]. Overexpression of this gene are known to be related to higher pathological tumour grade and clinical stage in PCa, leading to a higher risk of metastasis[Citation17]. The angiogenesis gene identified (ADM) is known to be expressed in PCa as a mitogenic factor that stimulates growth of cancer cell types and indirectly suppressed immune response, again leading to a higher risk of a metastatic event[Citation28]. Our findings suggest that dysregulation of these genes in PCa carcinogenesis may be in part mediated by aberrant DNA methylation, although future work is needed to clarify if these truly reflect aberrant methylation or SNP signals.
Our findings related to stem cell genes are novel. Several genes overexpressed in human embryonic stem cells are known to promote tumour cell migration, invasion, and proliferation [Citation75,Citation79]. Among studies that examined clinical outcomes, tumours from patients with relapse or death expressed elevated levels of embryonic stem cell markers compared to tumours in patients who were alive or who had not relapsed[Citation19]. In PCa patients, tumour expression of putative stem cell markers has been associated with bone metastasis[Citation20]. It has been suggested that higher expression of stem cells reflects a higher frequency of these cells in tumours and is thereby associated with worse clinical outcomes. DNA methylation plays a key role in maintaining the status of stem cell differentiation and silencing of these genes in differentiated cells [Citation79,Citation80]. If this hypothesis is correct, we would expect that DNA methylation profiles of stem cell identity will be predictive of PCa metastasis. Our findings support this hypothesis and urge future studies to evaluate the role of stem cell markers in clinical risk prediction models for PCa metastasis.
This study has several limitations. First, our algorithm may not have captured all metastases developed among untreated men, and some metastases could remain undiagnosed. Second, there was a large attrition of study subjects for obtaining methylation data (only 60% of the confirmed cases and 31% of the controls contributed to DNA methylation data). This was partly due to the retrospective nature of the study and, in part, due to the nature of small tumour size in the biopsy cores for those with very limited disease. Therefore, there is a potential selection bias, in that those with more ‘extensive’ disease may have been more likely to be included in the final analytical cohort. Finally, although our study intended to identify methylation markers to help inform treatment decisions, the use of DNA methylation markers in diagnostic biopsy may not be applicable to men with a very small tumour tissue in their diagnostic biopsy for whom sufficient tumour DNA for the methylation assay cannot be obtained.
This study has several unique strengths. First, our study design is innovative in that it focuses on untreated men with localized PCa to study the risk of disease progression without the influence of PCa treatment, uses diagnostic biopsy specimens to assess the tumour DNA methylome without surgery, and focuses on the most clinically relevant outcome (PCa metastasis). To our knowledge, this study is among the first to perform genome-wide methylation profiling using diagnostic biopsy PCa tissue to evaluate the prognostic value of DNA methylation markers for PCa metastasis in untreated patients. Second, our study is novel in that it examined the prognostic utility of stem cell markers, which has not been extensively studied.
Conclusion
We found that the DNA methylation status of several selected candidate genes was significantly associated with the risk of metastasis in patients with localized PCa who did not receive curative treatment after adjusting for known clinical prognostic characteristics. Future cohort studies of men undergoing active surveillance are needed to further validate our findings and evaluate whether these methylation markers could improve risk prediction to confirm the clinical utility of these markers. The candidate genes identified in this study may also have implications in the development of treatment strategies for PCa metastasis.
Data availability
Anonymized data that support the findings of this study may be made available from the corresponding author upon reasonable request from qualified researchers with documented evidence of training for human subjects protections.
Ethical approval
This study was approved by the KPSC Institutional Review Board and conducted in accordance with the U.S. Common Rule guidelines.
Supplemental Figure 1.docx
Download MS Word (384 KB)Supplemental Figure 3.docx
Download MS Word (70.1 KB)Supplementary Table 1.xlsx
Download MS Excel (764 KB)Supplemental Figure 2.docx
Download MS Word (190 KB)Acknowledgments
The authors thank the patients of Kaiser Permanente for helping us improve care through the use of information collected through our electronic health record system. This study was supported by a grant from the Department of Health and Human Services, National Institutes of Health, National Cancer Institute (5R01CA181242-05 [Chao]).
Disclosure statement
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Chao CR received research funding from Merck & Co. for research projects unrelated to this work. Slezak JM received research funding from Pfizer, Dynavax Technologies Corp., and ALK for projects unrelated to this work. Huang J is a consultant for or owns shares in the following companies: Amgen, Artera, Kingmed Diagnostics, MoreHealth, OptraScan, York Biotechnology, Genecode, Seagen Inc., and Sisu Pharma, and received grants from Zenith Epigenetics, BioXcel Therapeutics, Inc., Dracen Pharmaceuticals, and Fortis Therapeutics.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2308920
Additional information
Funding
References
- American Cancer Society. Cancer facts & statistics. 2021 [cited 2021 Sept].
- Li J, Siegel DA, King JB. Stage-specific incidence rates and trends of prostate cancer by age, race, and ethnicity, United States, 2004-2014. Ann Epidemiol. 2018 May;28(5):328–13.
- Slezak JM, Den Eeden SK V, Cannavale KL, et al. Long-term follow-up of a racially and ethnically diverse population of men with localized prostate cancer who did not undergo initial active treatment. Cancer Med. 2020 Nov;9(22):8530–8539.
- American Urological Association. Clinically localized prostate cancer: AUA/ASTRo/SUO guidelines. 2017 [cited 2021 Apr].
- Lam D, Clark S, Stirzaker C, et al. Advances in prognostic methylation biomarkers for prostate cancer.Cancers (Basel). 2020 Oct 15;12(10):2993. doi: 10.3390/cancers12102993
- Rodenhiser DI. Epigenetic contributions to cancer metastasis. Clin Exp Metastasis. 2009;26(1):5–18.
- Huynh KT, Hoon DS. Epigenetics of regional lymph node metastasis in solid tumors. Clin Exp Metastasis. 2012;29(7):747–756.
- Cooper CS, Foster CS. Concepts of epigenetics in prostate cancer development. Br J Cancer. 2009;100(2):240–245.
- Newell-Price J, Clark AJ, King P. DNA methylation and silencing of gene expression. Trends Endocrinol Metab. 2000;11(4):142–148.
- Li Q, Chen H. Epigenetic modifications of metastasis suppressor genes in colon cancer metastasis. Epigenetics. 2011 Jul;6(7):849–852.
- Shames DS, Minna JD, Gazdar AF. DNA methylation in health, disease, and cancer. Curr Mol Med. 2007;7:85–102.
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313–319.
- Okamoto OK. Cancer stem cell genomics: the quest for early markers of malignant progression. Expert Rev Mol Diagn. 2009;9(6):545–554.
- Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and caner stem cells. Nature. 2001;414(6859):6859):105–11.
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768.
- Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–323. doi: 10.1016/j.stem.2007.06.002
- Klarmann GJ, Hurt EM, Mathews LA, et al. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis. 2009;26(5):433–446. doi: 10.1007/s10585-009-9242-2
- Glinsky GV. “Stemness” genomics law governs clinical behavior of human cancer: implications for decision making in disease management. J Clin Oncol. 2008;26(17):2846–2853.
- Schoenhals M, Kassambara A, De Vos J, et al. Embryonic stem cell markers expression in cancers.Biochem Biophys Res Commun. 2009 May 29;383(2):157–162. doi: 10.1016/j.bbrc.2009.02.156
- Colombel M, Eaton CL, Hamdy F, et al. increased expression of putative cancer stem cell markers in primary prostate cancer is associated with progression of bone metastases. Prostate. 2012;72(7):713–729. doi: 10.1002/pros.21473
- Bastian PJ, Ellinger J, Wellmann A, et al. Diagnostic and prognostic information in prostate cancer with the help of a small set of hypermethylated gene loci. Clin Cancer Res. 2005;11(11):4097–4106.
- Yoon HY, Kim SK, Kim YW, et al. Combined hypermethylation of APC and GSTP1 as a molecular marker for prostate cancer: quantitative pyrosequencing analysis. J Biomol Screen. 2012;17(7):987–992.
- Phé V, Cussenot O, Rouprêt M. Methylated genes as potential biomarkers in prostate cancer. BJU Int. 2010;105(10):1364–1370.
- Chao CR, Slezak J, Siegmund K, et al. Genome-wide methylation profiling of diagnostic tumor specimens identified DNA methylation markers associated with metastasis among men with untreated localized prostate cancer. Cancer Med. 2023 Sep;12(18):18837–18849.
- Li LC. Epigenetics of prostate cancer. Front Biosci. 2007;12(8–12):3377–3397.
- Li Z, Wang C, Prendergast GC, et al. Cyclin D1 functions in cell migration. Cell Cycle. 2006;5(21):2440–2442.
- Gyorffy B, Lanczky A, Eklund AC, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–731. doi: 10.1007/s10549-009-0674-9
- Bertucci F, Birnbaum D. Distant metastasis: not out of reach any more. J Biology. 2009;8(3):28.
- Glinsky GV. Genomic models of metastatic cancer: functional analysis of death-fromcancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated Polycomb Group (PcG) protein chromatin silencing pathway. Cell Cycle. 2006;5(11):1208–1216.
- Ju HX, An B, Okamoto Y, et al. Distinct profiles of epigenetic evolution between colorectal cancers with and without metastasis. Am J Pathol. 2011;178(4):1835–1846. doi: 10.1016/j.ajpath.2010.12.045
- Iiizumi M, Liu W, Pai SK, et al. Drug development against metastasisrelated genes and their pathways: a rationale for cancer therapy. Biochim Biophys Acta. 2008;1786(2):87–104.
- Rinker-Schaeffer CW, Welch DR, Sokoloff M. Defining the biologic role of genes that regulate prostate cancer metastasis. Current Opin Urol. 2000;10(5):397–401.
- Hurst DR, Welch DR. Unraveling the enigmatic complexities of BRMS1-mediatedmetastasis suppression. FEBS Lett. 2011;585(20):3185–3190.
- Albini A, Mirisola V, Pfeffer U. Metastasis signatures: genes regulating tumor-microenvironment interactions predict metastatic behavior. Cancer Metastasis Rev. 2008;27(1):75–83.
- Keller ET, Fu Z, Brennan M. The biology of a prostate cancer metastasis suppressor protein: raf kinase inhibitor protein. J Cell Biochem. 2005;94(2):273–278.
- Chang YW, Bean RR, Jakobi R. Targeting RhoA/Rho kinase and p21-activated kinase signaling to prevent cancer development and progression. Recent Pat Anticancer Drug Discov. 2009;4(2):110–124.
- Henrique R, Ribeiro FR, Fonseca D, et al. High promoter methylation levels of APC predict poor prognosis in sextant biopsies from prostate cancer patients. Clin Cancer Res. 2007;13(20):6122–6129. doi: 10.1158/1078-0432.CCR-07-1042
- Rosenbaum E, Hoque MO, Cohen Y, et al. Promoter hypermethylation as an independent prognostic factor for relapse in patients with prostate cancer following radical prostatectomy. Clin Cancer Res. 2005;11(23):8321–8325. doi: 10.1158/1078-0432.CCR-05-1183
- Braga EA, Kashuba M VI, AV L VI, et al. New tumor suppressor genes in hot spots of human chromosome 3: new methods of identification. Mol Biol. 2003;37(2):194–211. doi: 10.1023/A:1023381218481
- Zlobec I, Minoo P, Baker K, et al. Loss of APAF-1 expression is associated with tumour progression and adverse prognosis in colorectal cancer. Eur J Cancer. 2007;43(6):1101–1107. doi: 10.1016/j.ejca.2007.01.029
- Mehta HH, Gao Q, Galet C, et al. IGFBP-3 is a metastasis suppression gene in prostate cancer. Cancer Res. 2011;71(15):5154–5163. doi: 10.1158/0008-5472.CAN-10-4513
- Min Y, Ghose S, Boelte K, et al. C/EBP-delta regulates VEGF-C autocrine signaling in lymphangiogenesis and metastasis of lung cancer through HIF-1alpha. Oncogene. 2011;30(49):4901–4909.
- Martin P, Liu YN, Pierce R, et al. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am J Pathol. 2011 Jul;179(1):422–435.
- Bailey CL, Kelly P, Casey PJ. Activation of Rap1 promotes prostate cancer metastasis. Cancer Res. 2009;69(12):4962–4968.
- Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008 Sep;18(9):421–429.
- Thomassen M, Tan Q, Kruse TA. Gene expression meta-analysis identifies chromosomal regions and candidate genes involved in breast cancer metastasis. Breast Cancer Res Treat. 2009;113(2):239–249.
- Chen L, Li H, Liu W, et al. Olfactomedin 4 suppresses prostate cancer cell growth and metastasis via negative interaction with cathepsin D and SDF-1. Carcinogenesis. 2011;32(7):986–994. doi: 10.1093/carcin/bgr065
- Iwakuma T, Agarwal N. MDM2 binding protein, a novel metastasis suppressor. Cancer Metastasis Rev. 2012 Dec;31(3–4):633–640.
- Revelos K, Petraki C, Gregorakis A, et al. Immunohistochemical expression of Bcl2 is an independent predictor of time-to-biochemical failure in patients with clinically localized prostate cancer following radical prostatectomy. Anticancer Res. 2005;25(4):3123–3133.
- Pinto AE, Andre S, Silva G, et al. BCL-6 oncoprotein in breast cancer: loss of expression in disease progression. Pathobiology. 2009;76(5):235–242.
- Grubb RL, Deng J, Pinto PA, et al. Pathway biomarker profiling of localized and metastatic human prostate cancer reveal metastatic and prognostic signatures. J Proteome Res. 2009;8(6):3044–3054.
- Fingleton B. Molecular targets in metastasis: lessons from genomic approaches. Cancer Genomics Proteomics. 2007;4(3):211–221.
- Gopalkrishnan RV, Kang DC, Fisher PB. Molecular markers and determinants of prostate cancer metastasis. J Cell Physio. 2001;189(3):245–256.
- Nicolson GL, Nawa A, Toh Y, et al. Tumor metastasis associated human MTA1 gene and its MTA1 protein product: role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clin Exp Metastasis. 2003;20(1):19–24.
- Yoshida BA, Chekmareva MA, Wharam JF, et al. Prostate cancer metastasis-suppressor genes: a current perspective. Vivo. 1998;12(1):49–58.
- Banez LL, Sun L, van Leenders GJ, et al. Multicenter clinical validation of PITX2 methylation as a prostate specific antigen recurrence predictor in patients with post-radical prostatectomy prostate cancer. J Urol. 2010;184(1):149–156. doi: 10.1016/j.juro.2010.03.012
- Weiss G, Cottrell S, Distler J, et al. DNA methylation of the PITX2 gene promoter region is a strong independent prognostic marker of biochemical recurrence in patients with prostate cancer after radical prostatectomy. J Urol. 2009;181(4):1678–1685. doi: 10.1016/j.juro.2008.11.120
- Hance MW, Dole K, Gopal U, et al. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J Biol Chem. 2012;287(45):37732–37744.
- Teng Y, Ngoka L, Mei Y, et al. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J Biol Chem. 2012;287(13):10051–10059.
- Wai Wong C, Dye DE, Coombe DR. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int J Cell Biol. 2012;2012:340296.
- Chen Q, Massague J. Molecular pathways: vcam-1 as a potential therapeutic target in metastasis. Clin Cancer Res. 2012;18(20):5520–5525.
- TE MS, Colnaghi MI, Colnaghi MI. The 67 kDa laminin receptor as a prognostic factor in human cancer. Breast Cancer Res Treat. 1998;52(1–3):137–145.
- Lu X, Kang Y. Epidermal growth factor signalling and bone metastasis. Br J Cancer. 2010;102(3):457–461.
- Gravdal K, Halvorsen OJ, Haukaas SA, et al. Proliferation of immature tumor vessels is a novel marker of clinical progression in prostate cancer. Cancer Res. 2009;69(11):4708–4715.
- Ghilardi G, Biondi ML, Cecchini F, et al. Vascular invasion in human breast cancer is correlated to T–>786C polymorphism of NOS3 gene. Nitric Oxide. 2003;9(2):118–122.
- Das S, Tucker JA, Khullar S, et al. Hedgehog signaling in tumor cells facilitates osteoblast-enhanced osteolytic metastases. PloS One. 2012;7(3):e34374.
- Larrieu-Lahargue F, Welm AL, Bouchecareilh M, et al. Blocking fibroblast growth factor receptor signaling inhibits tumor growth, lymphangiogenesis, and metastasis. PLoS One. 2012;7(6):e39540.
- Tawadros T, Brown MD, Hart CA, et al. Ligand-independent activation of EphA2 by arachidonic acid induces metastasis-like behaviour in prostate cancer cells. Br J Cancer. 2012;107(10):1737–1744.
- Vanella L, Di Giacomo C, Acquaviva R, et al. The DDAH/NOS pathway in human prostatic cancer cell lines: antiangiogenic effect of L-NAME. Int J Oncol. 2011;39(5):1303–1310. doi: 10.3892/ijo.2011.1107
- Prud’homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012;3(9):921–939.
- Tsirmoula S, Dimas K, Hatziapostolou M, et al. Implications of pleiotrophin in human PC3 prostate cancer cell growth in vivo. Cancer Sci. 2012;103(10):1826–1832.
- Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010;21(1):41–48.
- Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64(6):1975–1986. doi: 10.1158/0008-5472.CAN-03-3972
- Woodson K, O’Reilly KJ, Ward DE, et al. CD44 and PTGS2 methylation are independent prognostic markers for biochemical recurrence among prostate cancer patients with clinically localized disease. Epigenetics. 2006;1(4):183–186. doi: 10.4161/epi.1.4.3530
- Assou S, Le Carrour T, Tondeur S, et al. A meta-analysis of human embryonic stem cells transcriptome integrated into a web-based expression atlas. Stem Cells. 2007;25(4):961–973. doi: 10.1634/stemcells.2006-0352
- Tj T Jr, Weisenberger DJ, Den Berg D V, et al. Low-level processing of illumina infinium DNA methylation beadarrays. Nucleic Acids Res. 2013 Apr;41(7):e90.
- Mehrotra DV, Adewale AJ. Flagging clinical adverse experiences: reducing false discoveries without materially compromising power for detecting true signals.Stat Med. 2012 Aug 15;31(18):1918–1930. doi: 10.1002/sim.5310
- Reinsch CH. Smoothing by Spline Functions. Numerische Math. 1967;10(1):177–183.
- Atkinson S, Armstrong L. Epigenetics in embryonic stem cells: regulation of pluripotency and differentiation. Cell Tissue Res. 2008;331(1):23–29.
- Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors.Nature. 2008 Jan 10;451(7175):141–146. doi: 10.1038/nature06534