
ABSTRACT
Oxidative stress and neuronal dysfunction caused by intracerebral haemorrhage (ICH) can lead to secondary injury. The m6A modification has been implicated in the progression of ICH. This study aimed to investigate the role of the m6A reader YTHDC2 in ICH-induced secondary injury. ICH models were established in rats using autologous blood injection, and neuronal cell models were induced with Hemin. Experiments were conducted to overexpress YTH domain containing 2 (YTHDC2) and examine its effects on neuronal dysfunction, brain injury, and neuronal ferritinophagy. RIP-qPCR and METTL3 silencing were performed to investigate the regulation of YTHDC2 on nuclear receptor coactivator 4 (NCOA4). Finally, NCOA4 overexpression was used to validate the regulatory mechanism of YTHDC2 in ICH. The study found that YTHDC2 expression was significantly downregulated in the brain tissues of ICH rats. However, YTHDC2 overexpression improved neuronal dysfunction and reduced brain water content and neuronal death after ICH. Additionally, it reduced levels of ROS, NCOA4, PTGS2, and ATG5 in the brain tissues of ICH rats, while increasing levels of FTH and FTL. YTHDC2 overexpression also decreased levels of MDA and Fe2+ in the serum, while promoting GSH synthesis. In neuronal cells, YTHDC2 overexpression alleviated Hemin-induced injury, which was reversed by Erastin. Mechanistically, YTHDC2-mediated m6A modification destabilized NCOA4 mRNA, thereby reducing ferritinophagy and alleviating secondary injury after ICH. However, the effects of YTHDC2 were counteracted by NCOA4 overexpression. Overall, YTHDC2 plays a protective role in ICH-induced secondary injury by regulating NCOA4-mediated ferritinophagy.
Introduction
Intracerebral haemorrhage (ICH) represents a devastating affliction of the central nervous system (CNS), characterized by a substantial mortality rate and generally poor prognosis [Citation1]. The primary injury inflicted directly by the haemorrhage itself along with the secondary damage instigated by blood cell toxicity, excitotoxicity, oxidative stress, and inflammation can lead to critical disability or even death [Citation2]. There is growing evidence hinting that ferroptosis triggered by iron overload might have a role in causing brain injury following ICH [Citation3,Citation4]. Utilizing an elderly mouse model of ICH, studies showed that interception of the microRNA-124/ferroportin pathway improved neuronal cell survival by mitigating ferroptosis [Citation5]. Nevertheless, the distinct mechanisms through which ferroptosis influences the progression of ICH remain definitively unexplored.
Excessive autophagy is potentially implicated in the brain damage induced by endoplasmic reticulum stress following ICH [Citation6]. Autophagy has the capability to degrade cellular ferritin, thus causing an escalation in unstable iron concentrations within the cells, which in turn triggers ferroptosis [Citation7]. Numerous studies have shown that a protein referred to as nuclear receptor coactivator 4 (NCOA4) found on the surface of autophagosomes can initiate ferritin degradation [Citation8]. NCOA4 identifies and directly binds to the ferritin heavy chain (FTH1), thereby ferrying iron-bound ferritin to the autophagosome for eventual lysosomal degradation and the consequent release of iron [Citation9]. Stimulating NCOA4-mediated ferritinophagy accelerates ferroptosis and exacerbates early injury from ischaemia-reperfusion in mice [Citation10]. Nonetheless, the role of NCOA4 in ICH is yet to be comprehensively elucidated and therefore, necessitates further exploration.
The role of N6-methyladenosine (m6A) modification, which is crucial in assorted biological processes, and its potential involvement in the onset and progression of ICH is under investigation [Citation11]. In the model of ICH, it has been observed that silencing of the m6A methyltransferase, namely methyltransferase-like 3 (METTL3), is known to regulate the levels of Glutathione Peroxidase 4 (GPX4) mRNA, thereby impeding the evolution of ferroptosis [Citation12]. The m6A reader, YTH domain containing 2 (YTHDC2), is shown to foster the expression of Sirtuin 3 (SIRT3) by diminishing the stability of Lysine Demethylase 5B (KDM5B). This process enhances mitochondrial metabolic reprogramming in the context of diabetic peripheral neuropathy [Citation13]. The m6A reader, YTH Domain Family Member 1 (YTHDF1), is pinpointed as a significant factor impacting the stability of BECN1 mRNA. Inhibition of YTHDF1 could stymie docosahexaenoic acid (DHA)-induced autophagy-dependent ferroptosis in hepatic stellate cells [Citation14]. Regardless, the function of m6A readers in the context of ferritinophagy-mediated ferroptosis in ICH still remains unspecified and calls for further research.
Based on these findings, we constructed an ICH rat model by autologous blood injection and a Hemin-induced cell model to investigate whether m6A readers mediated the stability of NCOA4 mRNA, thereby influencing ferritinophagy to alleviate secondary injury after ICH. Our study aimed to provide a theoretical basis for better understanding the pathogenesis of ICH and identifying potential therapeutic targets.
Materials and methods
Establishment and intervention of the ICH rat model
Sprague-Dawley (SD) adult male rats (280–300 g) were obtained from Hunan SJA Laboratory Animal Co. Ltd. These rats were kept under ideal laboratory conditions entailing a temperature range of 25 ± 2°C, humidity levels around 50 ± 5%, and alternating 12-hour cycles of light and darkness. The rats had unrestricted access to food and water. One week post-purchase, the rats were partitioned randomly into two groups, labelled Sham and ICH, each group comprising of 6 rats. After anaesthesia, rats in the ICH group had their femoral artery isolated, and 50 μL of blood was taken. The rats were then fixed, and within 10 min, 50 μL of autologous blood was injected into the right basal ganglia region (0.2 mm anterior fontanelle, 3 mm right lateral, 5.8 mm depth) at a rate of 5 μL/min using a disposable sterile syringe (1 mL, outer diameter 0.45 mm, No. WH0040, WoHong, China). After 10-minute needle retention, the needle was slowly withdrawn to prevent blood reflux [Citation15]. In contrast to the ICH group, the Sham group rats underwent identical surgical procedures, exempting only blood injection. In the subsequent instance, an additional 40 rats were split randomly to constitute 4 groups; these were Sham, ICH, ICH+oe-NC, and ICH+oe-YTHDC2, with each group accommodating 10 rats. Rats in the ICH+oe-NC and ICH+oe-YTHDC2 groups were anesthetized with 3% pentobarbital sodium and fixed in a stereotaxic instrument. Then, a microinjection syringe (10 μL, outer diameter 0.31 mm, No. 7653–01, Hamilton, China) was used to inject 5 μL of YTHDC2-overexpressed lentivirus or its control (titre: 1 × 109 TU/mL, Abiowell, China) into the lateral ventricle (1.0 mm anterior fontanelle, 2.0 mm right lateral, 3.5 mm depth) at a rate of 0.5 μL/min within 10 min. After needle retention for 10 min, the needle was slowly withdrawn to prevent blood reflux. After 5 days, ICH model construction was performed as before. This study was approved by the ethics committee of Jining Medical University (JNRM-2023-DW-039).
Behavior test
The evaluation of neuronal dysfunction in rats was conducted using a modified Neurological Severity Score (mNSS) three days post ICH modelling. The mNSS is an inclusive test designed to measure aspects such as movement, sensation, balance, and reflex responses. The dysfunction severity is quantified on a scale ranging from 0 to 18, with a higher score signifying greater neurological malfunctions [Citation16].
Brain water content detection and histological analysis
Following behavioural assessments, we euthanized the rats via the method of cervical dislocation. We then gathered 4 mm thick coronal brain tissues from around the acupuncture point. A portion of these tissues was preserved with 4% paraformaldehyde, then embedded in paraffin, and sectioned for various staining procedures: Hematoxylin and Eosin (H&E), Nissl, Immunofluorescence (IF), and Prussian blue. The H&E staining enabled us to observe the arrangement of cells, their morphological integrity, and the distribution of chromatin. Nissl staining was used to examine the distribution and shape of neuronal cells. We used IF staining to analyse the presence of YTHDC2 and neuronal nuclear antigen (NeuN) in neuronal cells. In addition, we also used IF staining to detect the marker IBA1 of microglia and the marker GFAP of astrocytes. The Prussian blue staining method helped us to study the iron deposition in the brain tissues. We promptly weighed a portion of the fresh brain tissues immediately after extraction and then dried them at 100°C for 24 h before re-weighing. We calculated the brain’s water content (%) using the formula: (wet weight – dry weight)/wet weight × 100% [Citation17]. The remaining brain tissues were reserved for further analyses.
Extraction and culture of primary neuronal cells
Within 24 h of birth, foetal SD rats (provided by Hunan SJA Laboratory Animal Co., Ltd, Changsha, China) were euthanized, and their entire bodies were sterilized in a bath of 75% ethanol. Under sterile conditions, cortical tissues from these rats were carefully isolated and then minced with scissors. The resulting tissue samples were then treated with 5 mL of 0.25% trypsin (product code: AWC0232, manufacturer: Abiowell, China) and allowed to digest at a controlled temperature of 37°C for a duration of 15 min. The digestion process was then halted via washing with PBS three times, with each wash followed by centrifugation at a speed of 300 g for 5 min, after which the supernatant was discarded. The remaining primary neuronal cells were cultivated in Neurobasal-A medium (product code: 10888022, manufacturer: Gibco, USA), enhanced with a 10% addition of foetal bovine serum (FBS, product code: 10099141, manufacturer: Gibco, USA), 2% B27 supplement, 2 mg/L of glutamine, and 1% penicillin-streptomycin cocktail (product code: SV30010, manufacturer: Beyotime, China). Finally, the cultures were preserved in a humidified incubator (model: DH-160I, manufacturer: SANTN, China) with controlled conditions of 37°C and 5% CO2.
Cell grouping and treatment
In the experiment, cell groupings included Control, Hemin, Hemin+oe-NC, Hemin+oe-YTHDC2, Hemin+oe-YTHDC2+Erastin, Hemin+sh-NC, Hemin+sh-METTL3, Hemin+oe-YTHDC2+oe-NC, and Hemin+oe-YTHDC2+oe-NCOA4. The Control group of neuronal cells was treated with physiological saline for 24 h. The Hemin group of neuronal cells was treated with 100 μM Hemin (16009-13-5, Sigma-Aldrich, USA) for 24 h [Citation18]. The Hemin+oe-NC and Hemin+oe-YTHDC2 groups of neuronal cells were transfected with oe-NC or oe-YTHDC2 and treated with 100 μM Hemin for 24 h. The Hemin+oe-YTHDC2+Erastin group of neuronal cells was transfected with oe-YTHDC2 and treated with 5 µM Erastin (E7781, Sigma, USA) for 8 h, followed by 100 μM Hemin for 24 h. The Hemin+sh-NC and Hemin+sh-METTL3 groups of neuronal cells were transfected with sh-NC or sh-METTL3 and treated with 100 μM Hemin for 24 h. The Hemin+oe-YTHDC2+oe-NC group of neuronal cells was transfected with oe-YTHDC2 and oe-NC and treated with 100 μM Hemin for 24 h. The Hemin+oe-YTHDC2+oe-NCOA4 group of neuronal cells was transfected with oe-YTHDC2 and oe-NCOA4 and treated with 100 μM Hemin for 24 h. In the NCOA4 mRNA stability evaluation experiment, neuronal cells from the Control, Hemin, Hemin+oe-NC, and Hemin+oe-YTHDC2 groups were treated with 5 mg/mL Actinomycin D (Act D, 50-76-0, Sigma-Aldrich, USA) for 0, 3, 6, and 9 h after the aforementioned treatments. The sequences of sh-METTL3, oe-YTHDC2, and oe-NCOA4 were shown in Supplementary Text 1.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from samples using the Trizol reagent (15596026, Thermo, USA) and then converted to cDNA using the mRNA reverse transcription kit (CW2569, CWBIO, China). qRT-PCR was performed on QuantStudio1 Real-Time PCR System (ABI, USA) with UltraSYBR Mixture (CW2601, CWBIO, China). The relative gene expression was assessed by the 2−ΔΔCt method with β-actin being the reference gene. The primer sequences were listed in .
Table 1. Primer sequences.
Western blot
Total proteins were extracted from samples using radioimmunoprecipitation assay (RIPA) lysis buffer (AWB0136, Abiowell, China). The proteins were isolated through sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto nitrocellulose (NC) membranes. After being blocked with 5% skimmed milk (AWB0004, Abiowell, China) for 1.5 h, NC membranes were incubated with primary antibodies at 4°C overnight followed by the secondary antibodies at room temperature for 1.5 h. NC membranes were incubated with Super ECL Plus detection reagent (AWB0005, Abiowell, China) for chemiluminescence imaging. The grey values of protein bands were obtained using Quantity One 4.6.6 (Bio-Rad Inc., USA), and the protein expressions were calculated using β-actin as an internal reference. The information of antibodies was exhibited in . All the original western blot images were shown in Supplementary File 1.
Table 2. The information of antibodies.
Flow cytometry
To detect lipid reactive oxygen species (ROS) levels in brain tissues and neuronal cells, the samples were processed in the following manner: Initially, they were digested with 0.25% trypsin to create cell suspensions. Following trypsin digestion, the cell suspensions underwent two washes with PBS. Post-washing, they were then immersed in 5 μM C11-BODIPY (MX5211-1 MG, MaokangBio, China) and incubated in a 37°C environment for 1 h. After this incubation period, cells were again washed and suspended in PBS, and the lipid ROS levels were subsequently measured via a flow cytometer (A00-1-1102, Beckman, USA).
For the assessment of mitochondrial membrane potential (MMP), additional steps were required. After the typical digestion and centrifugation processes, neuronal cells were treated with 0.5 mL of a specific JC-1 staining solution. They were then maintained in a 37°C cell culture incubator for a further 30 min. Subsequent to this incubation period, the neurons were washed and resuspended in a solution of JC-1 staining buffer, all set for MMP detection.
Biochemical detection
Samples of whole blood were left to sit at room temperature for 2 h before being centrifuged at a force of 1000 g in a 4°C environment for 15 min. This process was utilized to extract the supernatant for further investigation. Subsequently, the concentrations of malondialdehyde (MDA), iron (Fe2+), and glutathione (GSH) within both rat serum and neuronal cells were quantified by following the instruction manuals of MDA assay kits (A003–1, Nanjing Jiancheng Bioengineering Institute, China), iron assay kits (ab83366, Abcam, UK) and GSH assay kits (A006-2-1, Nanjing Jiancheng Bioengineering Institute, China).
Cell counting kit-8 (CCK-8) assay
The neuronal cells underwent digestion using 0.25% trypsin and were densified at 5 × 103 cells/well, subsequently being incubated at 37°C. After achieving adhesion, they were subjected to appropriate treatment for a duration of 48 h. Following this, the original medium was superseded by 100 μL of medium, which contained a 10% concentration of the CCK-8 reagent (NU679, DOJINDO, Japan). In the concluding stage, the neuronal cells were cultivated over a 4-hour period, post which, the optical density (OD) at 450 nm was documented utilizing a multifunctional microplate reader (MB-530, HEALES, China).
RNA immunoprecipitation-qPCR (RIP-qPCR) assay
The RIP-qPCR assay was utilized to validate the interaction between YTHDC2 and NCOA4 and to measure the m6A levels of NCOA4 mRNA. Neuronal cells from each group were treated following the instructions provided by the Imprint® RNA Immunoprecipitation (RIP) Kit (RIP, Sigma, USA). The RNA was then purified and subjected to qPCR analysis.
Statistical analysis
The experimental data were analysed using GraphPad Prism 8.0 (GraphPad Software Inc., USA) and expressed as mean ± standard deviation of three independent experiments. The measurement data followed a normal distribution and showed homogeneity of variance, which was assessed using the Kolmogorov-Smirnov test and exploratory descriptive statistics test. The unpaired Student’s t-test was used for comparing the two groups, while one-way analysis of variance (ANOVA) and Tukey’s post-hoc test were applied for comparing multiple groups. Statistical significance was considered at a threshold of p < 0.05. All experiments were randomized and analysed blindly to minimize experimental bias.
Results
ICH changed the expression pattern of m6A readers in rats
An examination was conducted to determine the influence of ICH on the expression of m6A reader in rat cerebral tissues, analysed at both mRNA and protein levels using qRT-PCR and Western blot. Results demonstrated a notable reduction in the expressions of YTHDF1, YTHDF2, YTHDC2, YTHDF3, IGF2BP2, and YTHDC1 at mRNA and protein levels in the cerebral tissues of rats affected by ICH, when compared to the Sham group. Significantly, YTHDC2 displayed the most dramatic alteration. In contrast, the expressions of IGF2BP1 and IGF2BP3 remained unchanged. Consequently, YTHDC2 was selected for more comprehensive scrutinization (depicted in ). Due to the presence of bleeding in the striatum of the brain in the ICH rat model, we used IF staining to detect the rat striatum. It was confirmed that the expression of YTHDC2 and NeuN in neuronal cells of ICH rats was reduced compared to the Sham group (). In the ICH group, there is the higher expression of GFAP and IBA1 than in the Sham group. Moreover, there appears to be more co-localization of GFAP and YTHDC2, as well as IBA1 and YTHDC2, in the ICH group compared to the Sham group (). The above findings illustrated that the expression pattern of m6A readers was altered in ICH rats, particularly YTHDC2.
Figure 1. ICH altered the expression pattern of m6A readers in the brain tissues of rats. (a,b) The expressions of YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, IGF2BP1, IGF2BP2, and IGF2BP3 in brain tissues of rats were analyzed by qRT-PCR and Western blot. n = 3. (c) The expressions of YTHDC2 and NeuN in the neuronal cells of rats were identified by if staining. n = 3. (d) The expressions of YTHDC2 and GFAP in the astrocytes of rats were identified by if staining. n = 3. (e) The expressions of YTHDC2 and IBA1 in the microglia of rats were identified by if staining. n = 3. *p < 0.05 vs. Sham.
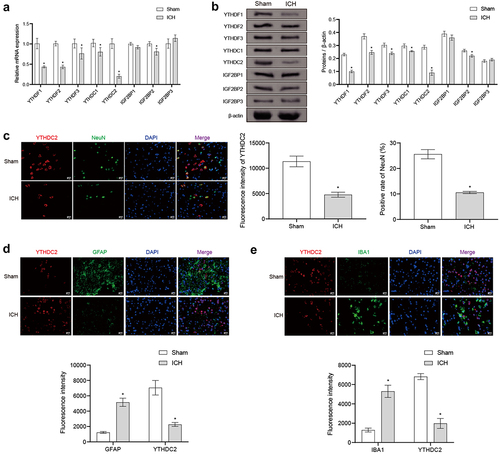
Regulation of YTHDC2 affected brain injury in ICH rats
To explain the role of YTHDC2 in ICH, an overexpression intervention of YTHDC2 was performed on ICH rats. Western Blot was used to detect whether oe-YTHDC2 is successfully overexpressed in brain tissues (Supplementary Figure S1A). YTHDC2 overexpression noticeably reduced the high mNSS score in ICH rats (). Further analysis revealed that YTHDC2 overexpression resulted in a decrease in the water content of the ipsilateral brain caused by ICH. However, neither ICH nor YTHDC2 overexpression had a significant effect on the water content of the contralateral brain and cerebellum (). H&E staining demonstrated that in the Sham group, the arrangement of neuronal cells was organized, with dense cytoplasm and intact nuclei. In the ICH and ICH+oe-NC groups, the arrangement of neuronal cells was more complex, with loose cytoplasm and condensed nuclei. Conversely, in the ICH+oe-YTHDC2 group, neuronal cells exhibited a relatively neat arrangement, with dense cytoplasm and relatively intact nuclei (). Nissl staining showed that in the Sham group, the brain tissue of rats had normal neuronal cell morphology with a large number of cells. In the ICH and ICH+oe-NC groups, the brain tissue showed injured neuronal cells with a significant number of cell deaths. In the ICH+oe-YTHDC2 group, the brain tissue showed alleviated neuronal cell injury with a relatively higher cell count (). These results demonstrated that regulation of YTHDC2 affected brain injury in ICH rats.
Figure 2. Regulation of YTHDC2 affected brain injury in ICH rats. (a) mNSS score. n = 10. (b) The water content of the contralateral brain, ipsilateral brain, and cerebellum. n = 3. (c The morphological changes in brain tissues were observed by H&E staining. n = 3. (d) The distribution and morphology of neuronal cells were analyzed by Nissl staining. n = 3. *p < 0.05 vs. Sham, #P < 0.05 vs. ICH+oe-NC.
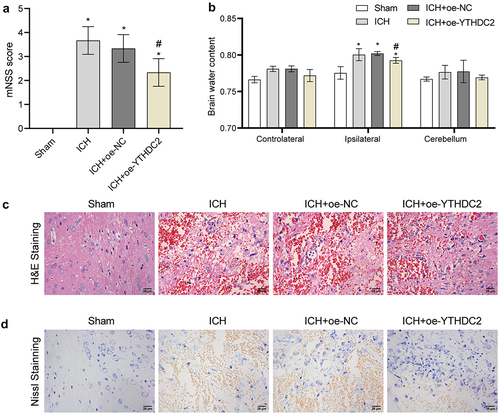
YTHDC2 affected ferroptosis and expression of ferritinophagy-related protein NCOA4 in ICH rats
To further clarify the regulatory role of YTHDC2 in brain injury, we assessed the levels of ferroptosis and ferritinophagy in neuronal cells of rats. Through Prussian blue staining, we observed a significant build-up of iron in the brain tissues of rats with ICH (intracerebral haemorrhage). However, we found that overexpression of YTHDC2 effectively reduced the iron accumulation caused by ICH (). Additionally, we observed elevated levels of lipid ROS (reactive oxygen species) in the brain tissues of ICH rats. Yet, YTHDC2 overexpression countered the promoting effect of ICH on ROS production (). Additionally, ICH resulted in a substantial increase in MDA and Fe2+ levels in the rat serum, while the synthesis and release of GSH were inhibited. However, these changes were reversed after YTHDC2 overexpression (). Furthermore, ICH upregulated the expressions of NCOA4, PTGS2, and ATG5 in the brain tissues of rats while downregulating the expressions of FTH and FTL. However, YTHDC2 overexpression partially counteracted the effects of ICH on the expression of these proteins (). These results displayed that YTHDC2 affected ferroptosis and expression of ferritinophagy-related protein NCOA4 in ICH rats.
Figure 3. YTHDC2 affected ferroptosis and expression of ferritinophagy-related protein NCOA4 in ICH rats. (a) Iron deposition in brain tissues was detected by Prussian blue staining. n = 3. (b,c) Lipid ROS levels in brain tissues were analyzed by flow cytometry. n = 4. (d,e) Levels of MDA, GSH, and Fe2+ in serum were measured by biochemical kits. n = 4. (f) The expressions of NCOA4, FTH, FTL, PTGS2, and ATG5 in brain tissues were assessed by Western blot. n = 4. *p < 0.05 vs. Sham, #P < 0.05 vs. ICH+oe-NC.
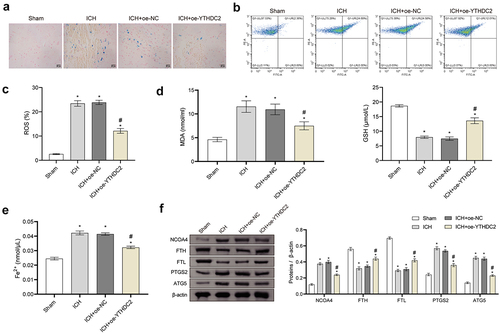
YTHDC2 affected neuronal injury through ferritinophagy-related ferroptosis
Based on the above data, we further investigated the mechanism of YTHDC2 on the neuronal injury. After the primary neuronal cells are extracted, immunofluorescence (IF) is used to detect the neuronal markers NeuN and MAP2 (Supplementary Figure S2). Hemin was used in neuronal cells to simulate an ICH model, and YTHDC2 overexpression and ferroptosis inducer, Erastin, were used for intervention. Western Blot was used to detect whether oe-YTHDC2 is successfully overexpressed in neuronal cells (Supplementary Figure S1(B)). The viability of neuronal cells decreased after Hemin treatment. However, YTHDC2 overexpression rescued the loss of cell viability, which was reversed by Erastin (). Moreover, YTHDC2 overexpression reduced the high levels of ROS induced by Hemin in neuronal cells. However, the beneficial effects of YTHDC2 overexpression were inhibited by the use of Erastin (). Furthermore, the use of Erastin counteracted the inhibition of YTHDC2 overexpression on the decrease of mitochondrial membrane potential (MMP) induced by Hemin ()). Additionally, after the use of Erastin, the inhibition of YTHDC2 overexpression on the production of MDA and Fe2+ and the promotion of GSH synthesis and release were significantly reversed ()). YTHDC2 overexpression inhibited the upregulation of NCOA4, PTGS2, and ATG5 expression induced by Hemin, as well as the downregulation of FTH and FTL expression. However, under the influence of Erastin, the effects of YTHDC2 overexpression on the expression of these proteins were partially counteracted ()). These results proved that YTHDC2 affected neuronal injury through ferritinophagy-related ferroptosis.
Figure 4. YTHDC2 affected neuronal injury through ferritinophagy-related ferroptosis. (a) The viability of neuronal cells was evaluated by CCK-8 assay. n = 3. (b,c) Levels of lipid ROS in neuronal cells were measured by flow cytometry. n = 3. (d,e) MMP in neuronal cells was determined by the JC-1 method. n = 3. (f,g) Levels of MDA, GSH, and Fe2+ in neuronal cells were evaluated by biochemical kits. n = 3. (h) The expressions of NCOA4, FTH, FTL, PTGS2, and ATG5 in neuronal cells were evaluated by Western blot. n = 3.*p < 0.05 vs. Control, #P < 0.05 vs. Hemin+oe-NC, &p < 0.05 vs. Hemin+oe-YTHDC2.
YTHDC2-mediated m6A modification regulated the mRNA stability of NCOA4
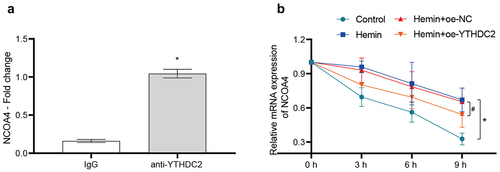
The above results indicated that YTHDC2 has regulatory control over the expression of NCOA4. Subsequently, we elaborated the distinct regulatory mechanism utilized by YTHDC2 on NCOA4. Evidence from the RIP-qPCR results confirmed the existence of an interaction between YTHDC2 and NCOA4 mRNA (). An assessment of the stability of NCOA4 mRNA revealed that its stability is increased in neuronal cells due to Hemin induction. Contrarily, the overexpression of YTHDC2 expedited the degradation of NCOA4 mRNA (). METTL3 is a core component of the m6A methyltransferase complex [Citation19]. To explore the role of YTHDC2-mediated m6A modification in the mRNA stability of NCOA4, METTL3 was silenced in neuronal cells (Supplementary Figure S3(a)). METTL3 silencing reduced the high m6A levels of NCOA4 mRNA induced by Hemin (Supplementary Figure S3(b)). These results indicated that YTHDC2-mediated m6A modification regulated the mRNA stability of NCOA4.
Figure 5. YTHDC2 bound to NCOA4 mRNA and affected its stability. (a) The binding of YTHDC2 and NCOA4 mRNA was verified by RIP-qPCR. n = 3.*p < 0.05 vs. IgG. (b) The stability of NCOA4 mRNA was analyzed by RT-qPCR. n = 3. *p < 0.05 vs. Control, #P < 0.05 vs. Hemin+oe-NC.
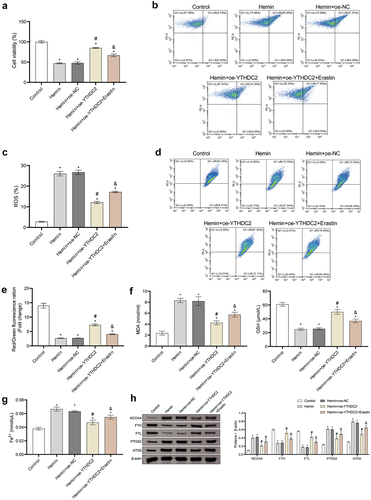
Regulation of NCOA4 reversed the effect of YTHDC2 on hemin-induced neuronal ferroptosis
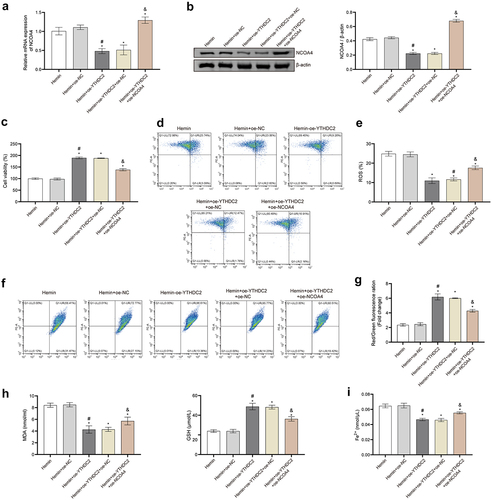
We undertook a comprehensive validation of the impact of YTHDC2 regulation on neuronal ferroptosis, via the overexpression of YTHDC2 and NCOA4. The upregulation of YTHDC2 resulted in the suppression of elevated NCOA4 expression, as induced by Hemin, discernible at both mRNA and protein levels. Conversely, overexpressing NCOA4 was found to reverse this pattern (). Furthermore, NCOA4 upregulation decidedly reduced the viability of oe-YTHDC2 transfected and Hemin-induced neuronal cells (). Notably, further examinations established that upregulating NCOA4 overturned the inhibitory effects of YTHDC2 upregulation on ROS production and MMP restoration in neuronal cells induced by Hemin (). The overexpression of YTHDC2 also led to the suppression of the accumulation of MDA and Fe2+ and to the promotion of GSH synthesis in Hemin-induced neuronal cells. However, these changes were found to be countered by the overexpression of NCOA4 (). Collectively, these results demonstrate the pivotal role of NCOA4 regulation in reversing the impact of YTHDC2 on neuronal ferroptosis triggered by Hemin.
Figure 6. Regulation of NCOA4 reversed the effect of YTHDC2 on hemin-induced neuronal ferroptosis. (a,b) The expression of NCOA4 in neuronal cells was detected by RT-qPCR and Western blot. n = 3. (c) The viability of neuronal cells was evaluated by CCK-8 assay. n = 3. (d,e) levels of lipid ROS in neuronal cells were analyzed by flow cytometry. n = 3. (f,g) MMP in neuronal cells was determined by the JC-1 method. n = 3. (h,i) Expressions of NCOA4, FTH, FTL, PTGS2, and ATG5 in neuronal cells were evaluated by Western blot. n = 3. *p < 0.05 vs. Hemin, #P < 0.05 vs. Hemin+oe-NC, &p < 0.05 vs. Hemin+oe-YTHDC2+oe-NC.
Discussion
ICH is a highly destructive form of stroke, characterized by significant incidence, mortality, and disability rates. It triggers cell death and consequentially results in secondary injuries [Citation20]. However, the precise mechanisms leading to these secondary injuries post-ICH remain a subject of study [Citation21]. Furthermore, the field currently lacks effective treatments to slow or halt the progression of the disease. This research, drawing on animal and cell experimentation, suggests that the m6A reader protein YTHDC2 might have the potential to counteract the secondary injuries following ICH. This effect may be achieved by the protein’s mediation of NCOA4 mRNA stability, which subsequently impacts ferritinophagy.
Injury to the CNS, including ICH, ranks as the predominant cause of both death and disability worldwide [Citation11]. Of recent, the phenomenon of m6A modification has deservedly captured the focused interest of researchers. This is largely due to its distinctive role in the orchestration of pathological and physiological processes through strategies such as splicing, translation, degradation, and the stabilization of RNA [Citation22]. Groundbreaking studies put forward that m6A modification is integral to brain development, establishing a significant link to a variety of neurological disorders, particularly those pertaining to CNS injuries [Citation23]. As it currently stands, m6A modification has demonstrated a noteworthy capacity to ameliorate neuronal dysfunction and suppress mechanisms such as cell apoptosis, necroptosis, ferroptosis, and inflammation within CNS injuries via various molecular pathways [Citation11]. Therefore, the m6A modification has shown significant potential as a target for therapeutic intervention in CNS injury. Proteins belonging to the YTH and IGF2BP families, which act as m6A readers, have the ability to bind to specific sites of RNA methylation, thereby influencing RNA metabolism and function [Citation24]. Recent studies have revealed the crucial role of YTHDF1 as a regulatory factor in cerebral ischaemia-reperfusion injury. By reducing the upstream stimulus factor 2 (Usf2), cerebral ischaemia-reperfusion injury can be alleviated through inhibition of YTHDF1-mediated translation of Cdc25A [Citation25]. Additionally, research has demonstrated that miR-421-3p can effectively suppress inflammation in cerebral ischaemia-reperfusion injury by targeting the m6A reader YTHDF1 and inhibiting the translation of p65 mRNA [Citation26]. These findings underscore the potential influence of the m6A reader YTHDF1 in cerebral ischaemia-reperfusion injury. Studies have also shown altered expression patterns of m6A readers following CNS injury. For instance, YTHDC1 was found to be induced after ischaemic stroke. Overexpression of YTHDC1 can promote the degradation of PTEN mRNA, thereby alleviating neuronal dysfunction [Citation27]. Here, the expression of YTHDC2 in brain tissues of ICH rats was remarkedly downregulated. However, the overexpression of YTHDC2 improved neuronal dysfunction, reduced brain water content, and decreased neuronal death in ICH rats. Thus, the m6A reader YTHDC2 has tremendous potential as a potential therapeutic target for ICH.
Iron is an essential trace element for maintaining the life of organisms, and iron imbalance can damage brain function due to the normal metabolism of the brain [Citation28]. After ICH, a large amount of iron is produced from haemoglobin degradation, and iron overload causes neurotoxicity and brain cell death [Citation29]. Ferroptosis is a novel form of cell death, which depends on the formation and accumulation of iron-mediated lipid peroxides. Studies have shown that ferroptosis of neuronal cells a critical role in ICH [Citation30]. In this study, the use of the ferroptosis inducer Erastin aggravated ferroptosis of neuronal cells induced by Hemin, manifested by an increase in ROS and Fe2+ levels, and a decrease in GSH levels. Recent studies have shown that ferroptosis inhibitor have neuroprotective effects on secondary injury after ICH. Inhibiting ferroptosis caused by iron overload and excessive accumulation of ROS can reduce brain injury and improve clinical outcomes in ICH patients [Citation31]. The ferroptosis inhibitor Ferrostatin-1 can alleviate neuronal dysfunction by reducing iron deposition in brain tissue surrounding the haematoma [Citation32]. Additionally, baicalin can alleviate motor deficits and brain injury in an ICH mouse model by inhibiting ferroptosis [Citation21]. Prior research indicates that elevated YTHDC2 expression may induce ferroptosis in LUAD cells, implying its role as a strong natural inhibitor of ferroptosis [Citation33]. Moreover, latest investigations reveal that the downregulation of YTHDC2 can effectively avert ferroptosis and deter the progression of hepatocellular carcinoma (HCC) in both in vitro and in vivo experiments [Citation34]. This present study found that overexpressing YTHDC2 significantly inhibited neuronal cell ferroptosis, thereby ameliorating secondary damage following an ICH. Hence, devising therapeutic interventions that target ferroptosis could potentially pave the way for innovative treatments for secondary ICH injuries.
Studies also suggest that autophagy can promote ferroptosis by modulating intracellular iron equilibrium and ROS production [Citation35]. Moreover, autophagy can break down ferritin, with NCOA4 recognized as the selective cargo receptor for ferritinophagy and responsible for ferritin degradation, leading to iron release [Citation36]. Li et al. have provided evidence confirming that a reduction in NCOA4 levels may potentially protect neurons from ferritinophagy-mediated ferroptosis [Citation37]. Additionally, Jin et al. further illustrated that in the model of cerebral ischaemia-reperfusion injury, the activation of NCOA4-mediated ferritinophagy can trigger oxidative stress, stimulate autophagy and cell apoptosis, eventually exacerbating brain damage [Citation10]. m6A modification is also significant in this process, with YTHDC1 purportedly targeting SQSTM1 in diabetic skin to regulate autophagy [Citation38]. Another research identified NCOA4 as an m6A-related ferroptosis gene [Citation39], suggesting that m6A modification may also be involved in ferritinophagy in the aftermath of ICH. Preliminary RIP-qPCR results from this study confirmed an interaction between YTHDC2 and NCOA4 mRNA. Further investigation suggested that YTHDC2 overexpression promotes NCOA4 mRNA degradation. However, this was reversed when there was an overexpression of NCOA4. It can be inferred that YTHDC2, as an m6A reader, inhibits NCOA4-mediated ferritinophagy by stabilizing NCOA4 mRNA – offering a possible pathway for reducing secondary ICH damage.
The current research focuses on the role and mechanism of m6A reader YTHDC2 in secondary injury after ICH. However, several limitations need to be addressed in future work. Firstly, the results of this study are obtained based on SD rats and neuronal cell models, which may not fully reflect the complex and dynamic characteristics of ICH in human patients. Therefore, it is necessary to include human samples or clinical data to enhance the credibility and persuasiveness of these findings. Secondly, this study primarily focuses on the impact of YTHDC2 on the regulation of neuronal dysfunction and ferritinophagy-related ferroptosis. However, it has not explored the potential roles of other cellular processes or molecular pathways that may be involved in secondary injury, such as inflammation or apoptosis. Further research is needed to comprehensively understand these pathways and their interactions with m6A modifications. Lastly, this study investigates the regulation of NCOA4 by YTHDC2 overexpression and its downstream effects. However, other approaches, such as YTHDC2 knockdown or targeted drug interventions, are not explored to further validate the research findings. By addressing these issues, the reliability and persuasiveness of the study can be further improved, and a better understanding of the role and mechanism of m6A reader YTHDC2 in secondary injury after ICH can be achieved.
Conclusion
In conclusion, our research highlights the vital role the m6A reader YTHDC2 plays in secondary injury after ICH. It was discovered that the diminished presence of YTHDC2 resulted in both neuronal dysfunction and additional brain injury in ICH cases. However, the overexpression of YTHDC2 was linked to improvements in the previously mentioned neuronal malfunction and a decrease in post-ICH brain trauma. Further, YTHDC2 was found to influence the stability of NCOA4 mRNA involved in ferritinophagy, which aids in mitigating secondary injury after an ICH episode. Such findings underscore the potential advantages of focusing therapeutic strategies on the m6A modification pathway for effective ICH treatment.
Supplementary Figure 3.jpg
Download JPEG Image (113.2 KB){kind=link}
Supplementary File 1.pdf
Download PDF (44.4 MB)Supplementary Figure 1.jpg
Download JPEG Image (223.5 KB){kind=link}
Supplementary Text 1.docx
Download MS Word (16.2 KB)Supplementary Figure 2.jpg
Download JPEG Image (1.5 MB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2326868
Additional information
Funding
References
- Hostettler IC, Seiffge DJ, Werring DJ. Intracerebral hemorrhage: an update on diagnosis and treatment. Expert Rev Neurother. 2019 Jul;19(7):679–15. doi: 10.1080/14737175.2019.1623671
- Xue M, Yong VW. Neuroinflammation in intracerebral haemorrhage: immunotherapies with potential for translation. Lancet Neurol. 2020 Dec;19(12):1023–1032. doi: 10.1016/S1474-4422(20)30364-1
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006 Jan;5(1):53–63. doi: 10.1016/S1474-4422(05)70283-0
- Zille M, Karuppagounder SS, Chen Y, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and Necroptosis. Stroke. 2017 Apr;48(4):1033–1043.
- Bao WD, Zhou XT, Zhou LT, et al. Targeting miR-124/Ferroportin signaling ameliorated neuronal cell death through inhibiting apoptosis and ferroptosis in aged intracerebral hemorrhage murine model. Aging Cell. 2020 Nov;19(11):e13235.
- Duan XC, Wang W, Feng DX, et al. Roles of autophagy and endoplasmic reticulum stress in intracerebral hemorrhage-induced secondary brain injury in rats. CNS Neurosci Ther. 2017 Jul;23(7):554–566.
- Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019 Oct 28;10(11):822.
- Mancias JD, Pontano Vaites L, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015 Oct 5;4. doi: 10.7554/eLife.10308
- Fang Y, Chen X, Tan Q, et al. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: a new mechanism of action. ACS Cent Sci. 2021 Jun 23;7(6):980–989. doi: 10.1021/acscentsci.0c01592
- Li B, Wang W, Li Y, et al. cGAS-STING pathway aggravates early cerebral ischemia-reperfusion injury in mice by activating NCOA4-mediated ferritinophagy. Exp Neurol. 2023 Jan;359:114269. doi: 10.1016/j.expneurol.2022.114269
- Tian M, Mao L, Zhang L. Crosstalk among N6-methyladenosine modification and RNAs in central nervous system injuries. Front Cell Neurosci. 2022;16:1013450. doi: 10.3389/fncel.2022.1013450
- Zhang L, Wang X, Che W, et al. Methyltransferase-like 3 silenced inhibited the ferroptosis development via regulating the glutathione peroxidase 4 levels in the intracerebral hemorrhage progression. Bioengineered. 2022 Jun;13(6):14215–14226.
- Jiao Y, Wang S, Wang X, et al. The m(6)A reader YTHDC2 promotes SIRT3 expression by reducing the stabilization of KDM5B to improve mitochondrial metabolic reprogramming in diabetic peripheral neuropathy. Acta Diabetol. 2023 Mar;60(3):387–399.
- Shen M, Guo M, Li Y, et al. m(6)A methylation is required for dihydroartemisinin to alleviate liver fibrosis by inducing ferroptosis in hepatic stellate cells. Free Radic Biol Med. 2022 Mar;182:246–259. doi: 10.1016/j.freeradbiomed.2022.02.028
- Sun J, Cai J, Chen J, et al. Krüppel-like factor 6 silencing prevents oxidative stress and neurological dysfunction following intracerebral hemorrhage via sirtuin 5/Nrf2/HO-1 axis. Front Aging Neurosci. 2021;13:646729. doi: 10.3389/fnagi.2021.646729
- Min LJ, Kobayashi Y, Mogi M, et al. Administration of bovine casein-derived peptide prevents cognitive decline in Alzheimer disease model mice. PLoS One. 2017;12(2):e0171515. doi: 10.1371/journal.pone.0171515
- Xiao L, Zheng H, Li J, et al. Targeting NLRP3 inflammasome modulates gut microbiota, attenuates corticospinal tract injury and ameliorates neurobehavioral deficits after intracerebral hemorrhage in mice. Biomed Pharmacother. 2022 May;149:112797. doi: 10.1016/j.biopha.2022.112797
- Yi X, Tang X. Exosomes From miR-19b-3p-Modified ADSCs Inhibit Ferroptosis in Intracerebral Hemorrhage Mice. Front Cell Dev Biol. 2021;9:661317. doi: 10.3389/fcell.2021.661317
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012 Apr 29;485(7397):201–6. doi: 10.1038/nature11112
- Ren S, Chen Y, Wang L, et al. Neuronal ferroptosis after intracerebral hemorrhage. Front Mol Biosci. 2022;9:966478. doi: 10.3389/fmolb.2022.966478
- Duan L, Zhang Y, Yang Y, et al. Baicalin inhibits ferroptosis in intracerebral hemorrhage. Front Pharmacol. 2021;12:629379. doi: 10.3389/fphar.2021.629379
- Wang Q, Liang Y, Luo X, et al. N6-methyladenosine RNA modification: a promising regulator in central nervous system injury. Exp Neurol. 2021 Nov;345:113829. doi: 10.1016/j.expneurol.2021.113829
- Yu J, She Y, Ji SJ. m(6)A modification in mammalian nervous system development, functions, disorders, and injuries. Front Cell Dev Biol. 2021;9:679662. doi: 10.3389/fcell.2021.679662
- Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014 Jan 2;505(7481):117–20. doi: 10.1038/nature12730
- Liu C, Gao Q, Dong J, et al. Usf2 deficiency promotes autophagy to alleviate cerebral ischemia-reperfusion injury through suppressing YTHDF1-m6A-Mediated Cdc25A translation. Mol Neurobiol. 2023 Nov 2. doi: 10.1007/s12035-023-03735-8
- Zheng L, Tang X, Lu M, et al. microRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A reader YTHDF1 to inhibit p65 mRNA translation. Int Immunopharmacol. 2020 Nov;88:106937. doi: 10.1016/j.intimp.2020.106937
- Zhang Z, Wang Q, Zhao X, et al. YTHDC1 mitigates ischemic stroke by promoting akt phosphorylation through destabilizing PTEN mRNA. Cell Death Dis. 2020 Nov 13;11(11):977. doi: 10.1038/s41419-020-03186-2
- Wei Y, Song X, Gao Y, et al. Iron toxicity in intracerebral hemorrhage: physiopathological and therapeutic implications. Brain Res Bull. 2022 Jan;178:144–154. doi: 10.1016/j.brainresbull.2021.11.014
- Garton T, Keep RF, Hua Y, et al. Brain iron overload following intracranial haemorrhage. Stroke Vasc Neurol. 2016 Dec;1(4):172–184.
- Sun Y, Li Q, Guo H, et al. Ferroptosis and iron metabolism after intracerebral hemorrhage. Cells. 2022 Dec 25;12(1):90. doi: 10.3390/cells12010090
- Chen J, Wang Y, Wu J, et al. The potential value of targeting ferroptosis in early brain injury after acute CNS disease. Front Mol Neurosci. 2020;13:110. doi: 10.3389/fnmol.2020.00110
- Chen B, Chen Z, Liu M, et al. Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res Bull. 2019 Nov;153:122–132. doi: 10.1016/j.brainresbull.2019.08.013
- Ma L, Zhang X, Yu K, et al. Targeting SLC3A2 subunit of system X(C)(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021 May 20;168:25–43.
- Li Y, Guo M, Qiu Y, et al. Autophagy activation is required for N6-methyladenosine modification to regulate ferroptosis in hepatocellular carcinoma. Redox Biol. 2024 Feb;69:102971. doi: 10.1016/j.redox.2023.102971
- Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016 Sep;26(9):1021–32.
- Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014 May 1;509(7498):105–9. doi: 10.1038/nature13148
- Li C, Sun G, Chen B, et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol Res. 2021 Dec;174:105933. doi: 10.1016/j.phrs.2021.105933
- Liang D, Lin WJ, Ren M, et al. m(6)A reader YTHDC1 modulates autophagy by targeting SQSTM1 in diabetic skin. Autophagy. 2022 Jun;18(6):1318–1337.
- Li D, Chen T, Li QG. Identification of a m(6)A-related ferroptosis signature as a potential predictive biomarker for lung adenocarcinoma. BMC Pulm Med. 2023 Apr 18;23(1):128.