
ABSTRACT
Non-coding 886 (nc886, vtRNA2–1) is the only human polymorphically imprinted gene, in which the methylation status is not determined by genetics. Existing literature regarding the establishment, stability and consequences of the methylation pattern, as well as the nature and function of the nc886 RNAs transcribed from the locus, are contradictory. For example, the methylation status of the locus has been reported to be stable through life and across somatic tissues, but also susceptible to environmental effects. The nature of the produced nc886 RNA(s) has been redefined multiple times, and in carcinogenesis, these RNAs have been reported to have conflicting roles. In addition, due to the bimodal methylation pattern of the nc886 locus, traditional genome-wide methylation analyses can lead to false-positive results, especially in smaller datasets. Herein, we aim to summarize the existing literature regarding nc886, discuss how the characteristics of nc886 give rise to contradictory results, as well as to reinterpret, reanalyse and, where possible, replicate the results presented in the current literature. We also introduce novel findings on how the distribution of the nc886 methylation pattern is associated with the geographical origins of the population and describe the methylation changes in a large variety of human tumours. Through the example of this one peculiar genetic locus and RNA, we aim to highlight issues in the analysis of DNA methylation and non-coding RNAs in general and offer our suggestions for what should be taken into consideration in future analyses.
Background
Non-coding 886 (nc886, HGNC symbol vtRNA2–1, previously also referred to as pre-miR-886, CBL3 and hvg-5) is the only known polymorphically imprinted gene in humans, the variation of which is not caused by genetic factors [Citation1–5]. In population cohorts of mainly European ancestry, 75% of individuals have been reported to present a maternally imprinted region in this locus (chr5:136078784–136080957, GRCh38 [Citation2]), while approximately 25% present two non-methylated alleles [Citation1,Citation6–8] (). A non-coding RNA is transcribed from the locus, but its nature has been discussed in the literature – i.e., whether the full-length RNA forms a hairpin structure and binds directly to proteins or is cleaved into miRNA-like short RNAs [Citation9,Citation10]. The expression of this RNA is regulated by the methylation pattern in the gene locus [Citation1,Citation11,Citation12], but also by the genetic variation downstream of the gene [Citation1].
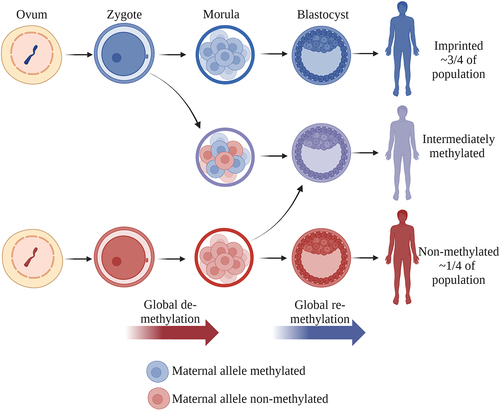
Figure 1. (a) Distribution of the median methylation levels of cg07158503, cg11608150, cg06478886, cg04481923, cg18678645, cg06536614, cg25340688, cg26896946, cg00124993, cg08745965 and cg18797653, located in the DMR overlapping nc886 (GSE40279). Two clusters can be observed: individuals with a 50% methylation level (~75% of the population) and those with a methylation level close to 0% (~25% of the population). In the former, the maternal allele is methylated [Citation6–8,Citation42,Citation43] and the paternal allele unmethylated and permissive for transcription, whereas in the latter cluster, both alleles are unmethylated and permissive for transcription. (b) Schematic presentation of the nc886 gene, nc886 DMR and the CTCF-binding sites flanking the DMR. The telomeric CTCF-binding site has been suggested to interact with another binding site near the IL9 gene, bringing a suggested enhancer region close to the nc886 gene [Citation1,Citation27]. Made with BioRender.com.
![Figure 1. (a) Distribution of the median methylation levels of cg07158503, cg11608150, cg06478886, cg04481923, cg18678645, cg06536614, cg25340688, cg26896946, cg00124993, cg08745965 and cg18797653, located in the DMR overlapping nc886 (GSE40279). Two clusters can be observed: individuals with a 50% methylation level (~75% of the population) and those with a methylation level close to 0% (~25% of the population). In the former, the maternal allele is methylated [Citation6–8,Citation42,Citation43] and the paternal allele unmethylated and permissive for transcription, whereas in the latter cluster, both alleles are unmethylated and permissive for transcription. (b) Schematic presentation of the nc886 gene, nc886 DMR and the CTCF-binding sites flanking the DMR. The telomeric CTCF-binding site has been suggested to interact with another binding site near the IL9 gene, bringing a suggested enhancer region close to the nc886 gene [Citation1,Citation27]. Made with BioRender.com.](/cms/asset/110930cd-0dac-44a1-b664-b262dcfc83c4/kepi_a_2332819_f0001_oc.jpg)
There are a number of studies showing how preconceptional or prenatal conditions associate with the methylation pattern of this gene [Citation1,Citation7,Citation8,Citation13,Citation14], and both the methylation pattern and RNAs expressed from this locus have been linked to health traits [Citation1,Citation7] and morbidities [Citation15,Citation16], making it a candidate for a molecule mediating the Developmental Origins of Health and Disease (DOHaD) hypothesis [Citation17]. However, due to the bimodal DNA methylation pattern of the nc886 locus and the considerable and stable physiological variation in the nc866 RNA expression, conservative analysis methods may produce false-positive or inconsistent results. Herein, we discuss why and how the characteristics of nc886 give rise to the contradictory results presented in current literature, in addition to providing new, reanalysed and replicated data. We pinpoint the unanswered questions regarding nc886 and emphasize the requirements for future studies regarding this genetic locus and the RNA(s) produced from it.
Discovery of nc886 RNA(s)
Non-coding 886 was first discovered as miR-886-3p and 886-5p by human short RNA sequencing [Citation18]. It was soon also identified in rhesus macaques [Citation19] and included in miRbase version 10. For an RNA to be considered as a miRNA with high confidence, it should produce a mature product of approximately 22 nt in length, have a hairpin-structured precursor and be phylogenetically conserved, and the decrease in Dicer enzyme function should lead to increased precursor molecule levels [Citation20]. These requirements were met by the nc886 primary transcript, which is cleaved into two short RNAs. In 2009, however, nc886 RNA was shown to have significant sequence similarities with vault-RNAs [Citation9,Citation21] and co-sediment with intact vault particles [Citation22], thus leading to the removal of miR-886 from miRbase (version 16). The RNA was suggested to be a novel vault RNA (vtRNA) [Citation23] and renamed as vtRNA2–1, which remains the HGNC-accepted official name of the gene. Like the three previously known vault RNAs (vtRNA1–1, 1–2 and 1–3), nc886 is coded in the 5q31, has a suitable length for a vault RNA, and has been suggested to form similar secondary structures to those formed by the other members of the vault family [Citation24].
Since then, the identity of the nc886 transcript both as a miRNA and as a vault RNA has been questioned. In 2011, Lee et al. recharacterized the nc886 RNA and concluded that the stem-loop of nc886 has qualities distinct from classical pre-miRNA and, unlike the majority of pre-miRNAs, its production is independent of Drosha. Dicer was also reported to cleave the stem-loop to two ~ 20nt RNAs with poor efficiency, and the levels of these short RNAs were shown to be so low that they could not be detected in the majority of the analysed samples [Citation25]. The nc886 RNA was also shown to be transcribed by RNA polymerase III (RNA pol III) [Citation26], while the majority of human miRNAs are transcribed by RNA polymerase II. On the other hand, it was demonstrated that the sequence similarity to the previously known vtRNAs is mostly limited to regions relevant to transcription by Pol III, that the expression pattern of nc886 differs from other vtRNAs and that vault proteins and the nc886 RNA do not to co-localize [Citation25]. The Dicer-dependent, but Drosha-independent, production of the small RNAs derived from this region was later verified by Miñones-Moyano et al. and Fort et al. [Citation9,Citation16]. However, Fort et al. demonstrated that the produced small RNAs associate with Argonaute, satisfying the attributes of miRNA, and further hypothesized that the suggested recent evolutionary origin of the nc886 gene would be the reason for the poor efficiency of Dicer in producing the mature miRNAs [Citation9,Citation27]. This work was again followed by the work of Lee et al., describing the issues arising from the classification of nc886 as a miRNA and further emphasizing their previous view of nc886 being distinct from pre-miRNAs [Citation10].
Regardless of the classification of nc886, the gene has been shown to code for a 101nt-long non-coding RNA, transcribed by Pol III [Citation26,Citation28], which is then cleaved into two short RNAs (nc886-3p; 23nt, and nc886-5p; 24-25nt) with rather low efficiency [Citation9,Citation25]. The full-length nc886 is highly expressed in at least cancer cell lines (105 copies per cell in HeLa cells), the transcripts are localized in the cytoplasm in an organized way [Citation25,Citation29], and the half-life of the transcripts has been reported to be short (71.5 min) [Citation12,Citation15,Citation25].
Function of nc886 RNAs
In the literature, the function of the nc886 RNAs has been suggested to be mediated either by the short RNAs acting as miRNAs [Citation1,Citation9] or by a direct binding of the hairpin structure to protein kinase R (PKR) (nucleotides 46–59) [Citation28,Citation30,Citation31,Citation44] or to 2–5-oligoadenylate synthetase 1 (OAS1) [Citation32]. Lee et al. have suggested a tumour-surveillance model where the binding of the nc886 RNA prevents PKR from being activated and suppresses apoptosis [Citation25,Citation33]. On the other hand, the short nc886 RNAs have been repeatedly predicted to regulate pathways linked with cancer development and insulin signalling [Citation1,Citation9]. It has also been suggested that the effects of the nc886 RNA would be at least partly caused by its binding to RISC and the subsequent hindering of the processing of other miRNAs [Citation29]. The expression levels of either the full-length nc886 RNA or the short derivates have been associated with infection [Citation23,Citation34–37], allergy [Citation38], asthma [Citation39] and skin senescence [Citation40,Citation41]. Since most of these studies argue for the existence of nc886-derived small RNAs based on arrays or TaqMan probes, the identity of the quantified RNAs must be interpreted with caution, as discussed by Lee et al. [Citation10]. All in all, consequences of nc886 RNA expression have been scarcely studied outside of cancer (reviewed later), and many open questions remain about its function in physiological conditions.
Methylation status of the nc886 locus
The nc886 gene is located in the only known canonically polymorphically imprinted region, the methylation status of which is not associated with genetics in humans [Citation1–3,Citation5]. In detail, there is an approximately 1,600-bp-long, differently methylated region (DMR), including the nc886 coding sequence, which is flanked by two CTCF binding sites that are hypothesized to insulate the DMR () [Citation2,Citation7]. Somatic diploid cells present each genomic site twice (maternal and paternal copies), and just as the methyl group in one DNA strand can be either present or lacking, the DNA methylation of a CpG site in one cell can be either 0%, 50% or 100%. When analysing samples with a mixture of cells, the DNA methylation of a given CpG site usually becomes a continuous variable, with values ranging from 0% to 100% (or from 0 to 1 in beta-values), as the sample is comprised of different proportions of cells with the previously mentioned DNA methylation statuses. However, the DMR overlapping the nc886 presents a mostly bimodal methylation pattern, where 75% of individuals have 50% methylation in the region (hereafter referred to as imprinted individuals) and approximately 25% of individuals have methylation levels close to 0% (hereafter referred to as non-methylated individuals), indicating two non-methylated alleles [Citation1,Citation6–8] (). Studies on family units [Citation42,Citation43] and gametes [Citation6–8,Citation43] strongly suggest that it is the maternal allele that is methylated in individuals with a 50% methylation level in this locus. It must be highlighted that the bimodal methylation pattern poses challenges for both data processing [Citation45] and analysis, as the majority of genome-wide analyses rely on linear regression methods and are thus based on the assumption that DNA methylation in a given site is a continuous, normally distributed variable.
Due to the parent-of-origin-dependent methylation pattern, nc886 is considered to be an imprinted gene. Canonically imprinted genes present a parent-of-origin-dependent gene expression pattern, with either the maternal or paternal locus silenced via epigenetic mechanisms, including DNA methylation [Citation46]. Typically, imprinted genes present a methylation level of 50% in all somatic cells and tissues [Citation47]. The epigenetic profiles maintaining the imprint are established during gametogenesis, when the existing DNA methylation pattern is first erased, followed by the creation of the parent-of-origin-related DNA methylation pattern [Citation48]. This pattern is then retained throughout an individual’s life. In addition, tissue- or developmental-stage-specific imprinting can be observed in, for example, the placenta [Citation49,Citation50]. The significance of intact genetic imprints is highlighted by the severe disorders caused by imprinting defects [Citation51]. We have previously shown that individuals who present multilocus imprinting disturbances also present an altered methylation pattern at the nc886 locus [Citation6], indicating that there are similarities in imprint establishment and/or maintenance of the nc886 imprint and the more typical non-polymorphically imprinted genes.
Similarly to canonically imprinted genes, individuals present the same methylation pattern of the nc886 DMR in the majority of their somatic tissues, regardless of the germ layer from which the tissue originates [Citation6,Citation8,Citation13]. We have previously reported that, among the 30 studied somatic tissues, only skeletal muscle and the cerebellum make an exception. In skeletal muscle, all individuals present an imprinted nc886 profile with a 50% methylation level, and, in the cerebellum, all individuals present a methylation level of approximately 75%, indicating biallelic methylation in some of the cells [Citation6]. Results from Olsen et al. indicate that the bimodal methylation pattern is also lost from granulosa cells and that the methylation of the nc886 locus in these specific cells might be associated with age [Citation52]. Upon analysing somatic tissues that were not included in the previous study, we now report that the methylation pattern of breast, testis and prostate tissues are also distinct from what can be observed in most somatic tissues, including blood (Supplementary Figure S1). Similarly to skeletal muscle, the bimodal methylation pattern cannot be observed in these tissues.
A small percentage of humans (1%–6%) present intermediate methylation levels (20%–40%) at the nc886 locus and are thus chimeric of non-methylated and imprinted cells. We have shown that the intermediate methylation pattern in blood is not due to different proportions of cell types [Citation1] and that it can also be observed across tissues [Citation6]. Like the non-methylated and imprinted status, the intermediate status is also stable through time [Citation1,Citation13]. Furthermore, a few individuals (~0.1% of the population) present methylation levels of over 60% [Citation1] (Supplementary Figure S2), indicating that the paternal allele has also gained methylation in some proportion of the cells, thus expressing very low levels of nc886 RNAs. This implies that at least certain cells can survive with very low nc886 expression.
nc886 is an evolutionarily young gene. It can only be found in primates, guinea pigs and some members of the squirrel family [Citation7,Citation27,Citation35]. In all of the apes analysed previously (n = 106), none presented a non-methylated epigenotype of nc886, indicating that the polymorphic imprinting of this locus is human-specific [Citation27]. The centromeric CTCF site, which is also evolutionarily young, is relevant for the existence of the methylation pattern in the region, as only primates with an intact binding sequence of CTCF present the nc886 imprint [Citation27].
Methylation of the nc886 locus is associated with nc886 RNA levels
The regulation of gene expression through DNA methylation is a nuanced system. The best-described mechanism is the association between repressed gene expression and methylated CpG islands overlapping the transcription start site of a gene [Citation53]. Methylation in the nc886 DMR has been shown to regulate nc886 RNA expression in several in vitro settings, including 5-Aza-2′deoxycytidine treatment [Citation11,Citation29,Citation54–56]. We and Treppendahl et al. have also shown that the intrinsic DNA methylation pattern in blood is associated with differences in nc886 expression levels [Citation1,Citation11]. In our data, the nc886-5p levels are increased two-fold in non-methylated individuals (both alleles permissive for expression) in comparison to imprinted individuals (only one allele permissive for expression). Furthermore, individuals presenting intermediate DNA methylation levels in the nc886 epiallele also present intermediate levels of nc886 RNAs [Citation1] (). Work by Park et al. describes how the methylation in the region leads to the formation of heterochromatin and the region being unavailable for RNA Pol III. They also suggest that, in open chromatin formation, MYC binds to an E-box upstream of the nc886 gene and then interacts with RNA Pol III, enabling transcription [Citation12]. Fort et al. have also demonstrated a negative correlation between the nc886 promoter methylation and chromatin accessibility in normal and tumour tissues [Citation37]. In addition to this epigenetic regulation of nc886 expression, we have shown that genetic variation 100–200 kb downstream of the nc886 gene is associated with nc886 RNA levels [Citation1]. As both the genetic profile and the DNA methylation pattern are stable in most somatic tissues and throughout an individual’s lifespan, there is already considerable physiological variation in nc866 RNA expression in healthy human population, which should be considered while investigating the levels of these RNAs in relation to morbidities.
Establishment of the nc886 imprint
Originally, Romanelli et al. suggested that the methylation pattern of nc886 arises 4–6 days into embryonic development [Citation43]. However, the results are based on cell lines derived from a limited number of individuals. A reanalysis of DNA methylation profiling data from the same cell lines (GSE52576) [Citation57] shows that the parthenogenetically activated oocytes and embryonic stem cells contain both non-methylated and imprinted cell lines (Supplementary Figure S3), indicating that these cell lines could actually be reflecting the intrinsic nc886 methylation pattern of the oocyte. Immortalization and the creation of iPSC have been shown to affect the methylation pattern of imprinted genes, including nc886, which should be taken into consideration when interpreting results from in vitro studies [Citation6,Citation12]. Our results with identical twins separated between days 1 and 3 [Citation6] after fertilization and the analysis of oocyte data by Carpenter et al. [Citation8] also suggest that the methylation pattern is already established in the oocyte. Although DNA methylation profiling data is currently available from oocytes in many studies, the coverage of the nc886 DMR in bisulphite sequencing data is generally so low that no definitive conclusion can be drawn. To some extent, this also includes the data of Okae et al. [Citation58] upon which the conclusions by Carpenter et al. [Citation8] and Jima et al. https://jb2.humanicr.org/ [Citation59] are based.
Although conclusive data are lacking, current results are pointing in the direction of the nc886 methylation pattern being established in the oocyte, in line with non-polymorphic maternally imprinted genes [Citation60]. In mice, these imprints are established asynchronously during the growth and maturation of the oocyte in prophase II [Citation61]. As the nc886 polymorphic imprint is unique to humans, the establishment of the methylation pattern is difficult to study because the growth phase of the oocyte lasts from the early weeks after the birth of the woman to the formation of the secondary follicle during menstruation, which can span up to 50 years. The establishment of the intermediate and over-methylated patterns, on the other hand, likely occurs during the de- and re-methylation of the embryonic genome, as identical twins separated before implantation can present methylation differences of up to 17% [Citation6]. This also fits the timing of the de- and re-methylation of the embryonic genome [Citation62] (). Thus, we suggest that there are at least two distinct mechanisms contributing to the nc886 methylation patterns observed in humans: the initial establishment of the pattern during oocyte maturation and the subsequent maintenance of this pattern during embryonic development.
Figure 2. Establishment of the nc886 methylation pattern. The methylation pattern of the non-methylated or imprinted nc886 locus is suggested to be established during the maturation of the oocyte. We further hypothesize that the intermediate methylation pattern is caused by the sporadic loss of methylation during the global de-methylation of the embryonic genome or the gain of methylation during the re-methylation. After implantation, the methylation pattern, as well as rough portions of non-methylated and imprinted cells in intermediately methylated individuals, remain unchanged in the majority of somatic tissues. Made with BioRender.com.
One of the burning questions relating to nc886 is what leads 25% of studied individuals to present the non-methylated epigenotype. Unlike with other polymorphically imprinted genes, targeted and genome-wide genetic analyses have failed to discover a genetic cause for the pattern [Citation1,Citation3,Citation8,Citation11,Citation13]. Based on 30,347 studied individuals from 32 datasets, we previously reported that in singletons of European ancestry, the proportion of imprinted individuals varies by less than 5%, ranging from 72.6% to 77.5%. When considering other ethnicities or populations consisting of twins, more variation could be observed [Citation6]. In our previous study, populations of African descent had a higher proportion of imprinted individuals (79.1% to 78.7%), whereas Asian populations included fewer imprinted individuals (68.2% to 65.8%), although the differences were not drastic and the number of populations of other than European ancestry was limited [Citation6]. A smaller percentage of imprinted individuals (65.9%, n = 82) in an Asian population is also described by You et al. [Citation63].
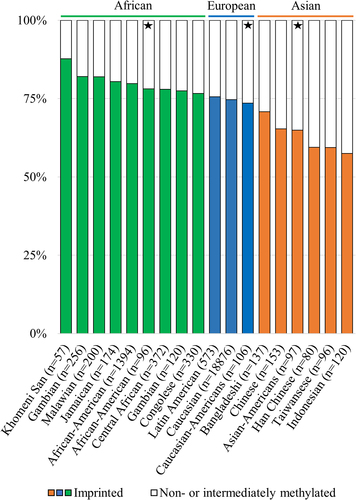
To provide more information on the topic, we investigated DNA methylation data from data repositories with a focus on populations of other than European ancestry and were able to replicate our previous findings (). When inspecting DNA methylation data from populations of African ancestry, the highest percentage of imprinted individuals observed was 88% in ǂKhomani San, albeit the sample population is limited in size (GSE99029, n = 57) [Citation64]. The pattern is similar among American populations of African descent [Citation65], as well as other African populations [Citation66]. On the other hand, the percentage of imprinted individuals is lower in Asian populations, and the lowest percentage of individuals with an imprinted nc886 was 55%, which was observed in populations from the Indonesian archipelago [Citation67] (). It should be noted that, within populations, differences can be seen between specific living locations. Therefore, we cannot rule out the possibility that local genetic or environmental factors could affect the percentage of individuals with an imprinted nc886 locus, but we also note that the observed differences could be explained by the small numbers of individuals in the specific sub-populations (Supplementary Figure S4). As the establishment of the methylation pattern has not been associated with genetic variation [Citation1–3,Citation5], it is perplexing that systematic variation in the percentage of imprinted individuals was observed across populations. These results again highlight the need for more EWAS and mQTL analyses to be performed in non-European populations [Citation68].
Figure 3. Percentages of imprinted (in colour) and non- and intermediately methylated individuals in the nc886 locus in population cohorts with ancestral origins in Africa (green), Europe (blue) and Asia (orange). The percentage of imprinted individuals in populations with ancestral origins in Africa is higher, and in populations originating from Asia lower, than the 75% previously reported populations of European ancestryCitation1. Populations marked with a star are from the same sample series (GSE36369), and these populations are thus free of technical or sample collection bias when compared to each other. Data processing and thresholds for imprinted individuals are presented in the supplementary materials and methods and in Supplementary Figure S2.
The prevalence of non-methylated individuals has been associated with maternal age [Citation1,Citation7,Citation69], the season of conception [Citation7,Citation13], maternal nutrition [Citation13] or folic acid supplementation [Citation70], family socioeconomic status [Citation1] and maternal alcohol consumption [Citation8]. Of these, only the association between maternal age and a higher prevalence of non-methylated children has been shown in more than one cohort [Citation7,Citation13]. We further aimed to replicate the association between the season of conception and the prevalence of imprinted individuals. In the dataset in which this was first reported, the analysis was performed with nc886 methylation as a continuous variable [Citation13]. Later, Carpenter et al. [Citation7] analysed the data as a categorical variable, but removed the intermediately methylated individuals as having an ‘inconclusive’ methylation pattern. We again reanalysed the data by dividing individuals into imprinted and others to avoid losing individuals in a cohort with a limited size (n = 120). While the finding that links nc886 methylation to the season of conception remains nominally significant, it is not replicated in a dataset with a similar study setting (GSE99863) (). All in all, more research with larger datasets is needed to understand if, and especially how, preconceptional or prenatal maternal traits associate with the nc886 methylation profile. It remains to be established whether these traits change the methylation status of the oocyte or embryo, or whether a non-methylated nc886 region is beneficial for the success of fertilization or the survival of the foetus in certain conditions.
Figure 4. Reanalysing and attempting to replicate findings linking the nc886 methylation pattern to (a) the season of conception [Citation13] (GSE59592 and GSE99863), (b) Postnatal (10.5061/dryad.k67kf) and periconceptional (GSE116379) exposure to famineCitation14, and (c) Parkinson’s disease (GSE165081, GSE145361 and GSE111629). (a) The original discovery by Silver et al. (GSE59592Citation13) associating lower levels of nc886 methylation to the season of conception remains statistically significant even after clustering individuals into imprinted and non-methylated or intermediately methylated (p = 0.049), but this finding is not replicated in the other Gambian population cohort available. The significant association between postnatal exposure to famine (b) and Parkinson’s disease (c) disappears when the nc886 methylation pattern is treated as a categorical variable and is not replicated in datasets with similar or related study settings. Data processing and thresholds for imprinted individuals are presented in supplementary materials and methods and in Supplementary Figure S2.
![Figure 4. Reanalysing and attempting to replicate findings linking the nc886 methylation pattern to (a) the season of conception [Citation13] (GSE59592 and GSE99863), (b) Postnatal (10.5061/dryad.k67kf) and periconceptional (GSE116379) exposure to famineCitation14, and (c) Parkinson’s disease (GSE165081, GSE145361 and GSE111629). (a) The original discovery by Silver et al. (GSE59592Citation13) associating lower levels of nc886 methylation to the season of conception remains statistically significant even after clustering individuals into imprinted and non-methylated or intermediately methylated (p = 0.049), but this finding is not replicated in the other Gambian population cohort available. The significant association between postnatal exposure to famine (b) and Parkinson’s disease (c) disappears when the nc886 methylation pattern is treated as a categorical variable and is not replicated in datasets with similar or related study settings. Data processing and thresholds for imprinted individuals are presented in supplementary materials and methods and in Supplementary Figure S2.](/cms/asset/f67d4bb7-b9a6-4109-9d12-370bf3cf52da/kepi_a_2332819_f0004_oc.jpg)
If the proportions of nc886 methylation status groups in a population are affected by mechanisms related to the establishment and maintenance of the imprint, but also to the chances of a live birth in certain conditions, it would explain how the nc886 methylation pattern in populations could be affected by perinatal or periconceptional conditions while being simultaneously already established in the maturing oocyte. Furthermore, the combined contribution of three distinct mechanisms (establishment, maintenance, and survival bias) might mask the potential genetic component in the pattern, thus explaining the differences between populations with different geological origins while simultaneously explaining the negative results observed in genetic analyses.
Adulthood traits affecting nc886 methylation status
The nc886 methylation levels in blood have been shown to be stable for up to 25 years of follow-up in adulthood [Citation1] and from childhood to adolescence [Citation13]. In contrast to these findings, a multitude of studies have reported, in cross-sectional settings, that methylation status in this locus is affected by outside exposures after birth or in adulthood, including post-natal famine [Citation14], adulthood exposure to pesticides or gases/fumes [Citation71], miner dust [Citation72], smoking [Citation73], wildfire-related fine particulate matter [Citation74], and long-term aircraft and railway noise [Citation75]. One explanation for these conflicting findings is that the qualities of nc886 methylation have not been taken into account and, in these analyses, the methylation in the locus has been treated as a continuous variable, which can lead to false-positive findings. We have now reanalysed the available dataset related to post-natal famine [Citation14] by categorizing individuals based on their nc886 methylation status. No difference was seen in the prevalence of imprinted individuals between those exposed to famine either pre- or postnatally and controls, when reanalysing the data as categorical (). It should be noted that we were only able to analyse the whole data, not the selected subset utilized in the work by Finer et al. [Citation14]. Unfortunately, other datasets in which the above-mentioned associations were discovered are not publicly available and thus could not be reanalysed with nc886 methylation treated as a categorical variable. Furthermore, some of the findings are based on a very limited number of individuals, and in the case of prenatal famine, for example, similar studies have not been able to replicate the associations [Citation76–78] (). Therefore, a reanalysis of the datasets and replication are needed to rule out false-positive findings.
nc886 and later-life health traits
When treated as a categorical variable, the nc886 methylation pattern has been associated with metabolic traits. We have shown that, in comparison to imprinted individuals, non-methylated individuals have higher insulin and glucose levels in childhood and adolescence, non-methylated boys have higher HDL and non-HDL cholesterol levels in childhood, and non-methylated small children present with higher adiposity [Citation1]. In line with this, van Dijk et al. showed that non-methylated children were at an increased risk of obesity, a trend which they replicated in a dataset consisting of 355 healthy young individuals (GSE73103) [Citation79]. The maternal imprinted nc886 locus has also been reported to be associated with an increased risk of preterm birth (n = 82) [Citation63], but this interesting discovery still needs to be replicated in a larger dataset.
Many publications report associations between nc886 methylation status and the risk of morbidities, while treating the methylation in this region as a continuous variable. Lower methylation of nc886 has been associated with an increased risk of orofacial clefts [Citation80], IgA nephropathy [Citation56] and Parkinson’s disease [Citation81]. When reanalysing the nc886 methylation pattern as a categorical variable, it is clear that, in the discovery cohort of IgA nephropathy (GSE72364, n = 12 [Citation81]), four out of the six controls present the non-methylated epigenotype and all of the six affected individuals are imprinted, generating the Δβ > 0.3 (Supplementary Figure S2). With such a small population, the probability of having one group consisting of only imprinted individuals is 18%, and nominally significant results would be obtained by having 3 out of 6 individuals be non-methylated in the other group. Similarly, in the data of Henderson et al. (GSE165083 [Citation81]), the noticeable but statistically non-significant difference in the nc886 methylation status groups between Parkinson’s disease cases and controls (imprinted vs others in the Chi-squared test p = 0.09, n = 28) is generated by 8 vs 12 imprinted individuals in each group (Supplementary Figure S2). Furthermore, no difference can be observed in the nc886 methylation patterns of Parkinson’s patients and controls in GSE145361 [Citation82], with 1,889 individuals, or in GSE111629, with 572 individuals [Citation6,Citation83] ().
Similar issues can be seen when reporting the associations of nc886 RNA expression and phenotypes in small sample settings. For example, Miñones-Moyano et al. report elevated nc886-5p levels in the brains of Parkinson’s disease patients in comparison to controls (n < 40 per setting). A clustered expression pattern can be observed in the amygdala, the frontal cortex and the substantia nigra of Parkinson’s patients, with patients in the motor stages of the disease ( in Miñones-Moyano et al. [Citation16]). As the nc886 RNA levels are strongly regulated by the methylation pattern, their results could be caused by the uneven distribution of imprinted and non-methylated individuals among cases and controls. In support of this, the aforementioned connection to Parkinson’s disease cannot be detected in the cerebellum, where the bimodal methylation pattern cannot be observed [Citation6,Citation16]. Similarly, it would be interesting to see whether the associations between nc886 RNA levels and IgA nephropathy would remain significant after taking into account the stable regulators of the RNA levels [Citation84]. A reanalysis with the methylation and genetic regulators included in the model would be warranted to confirm these results.
nc886 and cancer
Changes in nc886 methylation level and RNA levels are widely reported in cancer. nc886 RNAs have been found to be upregulated in cervical cancer [Citation85–87], breast cancer [Citation86,Citation88], high-grade bladder cancer [Citation89], high-grade prostate cancer [Citation90,Citation91], multiple myeloma [Citation92], endometrial cancer [Citation32] and renal carcinoma [Citation93], while downregulation has been reported in cholangiocarcinoma [Citation30], oesophageal cancer [Citation54,Citation94], prostate cancer [Citation95,Citation96], ovarian cancer [Citation97], breast cancer [Citation98], thyroid cancer [Citation99,Citation100], small-cell lung cancer [Citation101], squamous cell lung carcinoma [Citation102] and oral squamous cell carcinoma [Citation103]. Notably, in breast and prostate cancers, both directions are reported. Several molecular mechanisms could explain the changes in nc886 transcription in cancer. The expression of genes transcribed by RNA pol III, such as nc886, has been reported to be upregulated in cancer. The transcription of nc886 is also activated by transcription factor MYC, the levels of which are often also upregulated in cancerous cells [Citation12], theoretically leading to even higher expression rates of nc886 RNAs. On the other hand, hypermethylation of the nc886 locus has been reported in several types of cancer [Citation43,Citation55], leading to the formation of heterochromatin and the repression of the transcription [Citation12].
Knocking out or down nc886 RNA expression has been shown to both induce apoptosis or suppress proliferation [Citation25,Citation28,Citation44,Citation85–88,Citation91,Citation93,Citation104] and to promote cell division [Citation105]. Similarly, overexpression of these RNAs has been shown to have both growth-promoting [Citation85,Citation86,Citation91,Citation93,Citation106] and growth-restricting [Citation9,Citation54,Citation95,Citation99,Citation103,Citation107–109] properties. Elevated levels of the nc886 RNAs or lower levels of DNA methylation in the region have also been linked to both a poorer [Citation29,Citation91,Citation101,Citation110–112] and a better [Citation11,Citation54,Citation96,Citation105,Citation107,Citation108,Citation113,Citation114] prognosis of the malignant disease. The issues of investigating the roles of alleged miRNAs in cell cultures with over-expression or knock-out has been described in detail by Lee et al., emphasizing the selection of suitable methods to produce a natural nc886 molecule in physiological concentrations, in addition to correct methods in evaluating the success of knock-down [Citation10]. In the case of nc886, the stable methylation pattern and the genetic variation in the enhancer region lead to substantial intrinsic differences in nc886 RNA levels between individuals [Citation1]. These differences should be taken into account when studying associations between nc886 RNA levels and end points in small sample sets or in limited numbers of cell lines, as uneven distribution of the stable regulatory profiles in cases and controls may lead to false-positive findings.
Changes in the methylation pattern of the nc886 locus have also been linked to cancer [Citation37,Citation43,Citation55,Citation115]. However, the majority of these studies do not take into account the bimodal distribution of normal tissue or that the changes in the DNA methylation pattern lead into different consequences in relation to the original methylation status. If the methylation status increases by 20%, it will lead to very different results depending on whether the inherent methylation level was 0% or 50%. In studies where the original methylation pattern has been considered, both hyper- and hypomethylation have been reported [Citation43,Citation55,Citation115], with both also reported to have predicted poorer survival [Citation37,Citation115]. Also, the innate DNA methylation status of the nc886 locus has been shown to predict the future risk of prostate [Citation4,Citation5], breast [Citation5,Citation42] and pancreatic cancer [Citation116], although the study by Wang et al. treats nc886 methylation as a continuous variable and reports rather inconsistent results [Citation116]. These studies suggest that individuals with non-methylated nc886 might be at an increased risk of developing cancer, which would, at least in theory, be in line with the suggested growth-promoting role of maternally imprinted genes, such as nc886, during foetal development [Citation117].
Ignoring the special qualities of nc886 methylation can also hide biologically relevant results. As an example, when comparing the methylation levels in the region from clear cell renal cell carcinoma (GSE61441) and adjacent normal tissue, a statistically non-significant (p = 0.06) hypomethylation can be detected in the tumours. When comparing the within-individual differences between normal and tumour tissue, 65% (30/46) of the tumours show higher than 2% hypomethylation, while 13% (6/46) present hypermethylation of over 2%, suggesting a pattern of hypomethylation but also stark differences between individual tumours. Intriguingly, hypomethylation can also be seen in individuals presenting low methylation levels in healthy tissue ().
Figure 5. (a – c) The median methylation level of nc886 in tumour samples (in black) from the cancer genome atlas program (TCGA) and from corresponding healthy tissues (in grey). (a) In some cancer types, the original bimodal methylation pattern is faithfully or near-faithfully maintained (acute myeloid leukaemia and hepatocellular carcinoma), while in others, the pattern is completely lost (malignant melanoma). In tumours from the cerebrum (excluding glioblastoma), a systematic hypermethylation can be observed. (b) the bimodal methylation pattern of nc886 is already missing in the healthy breast, prostate and testis samples, with breast and prostate cancer demonstrating hypermethylation, whereas the pattern in testicular tumours corresponds to the hypomethylated unimodal pattern also observed in the healthy tissue. (c) interestingly, renal carcinomas are observed to present hypomethylation in comparison to healthy tissue, while some malignant rhabdoid tumours of the kidneys are hypermethylated. (d) when analysing median nc886 methylation from the healthy adjacent tissue and the tumour in clear cell adenocarcinoma (GSE61441)Citation135, a non-significant minor hypomethylation can be observed in the tumours, but an analysis of changes at the individual level reveals a more systematic hypomethylation pattern. The start of the arrow represents the methylation level of the healthy tissue and the end of the arrow the methylation level of the tumour, with each arrow representing one individual. Data processing and thresholds for imprinted individuals are presented in supplementary materials and methods and in Supplementary Figure S5.
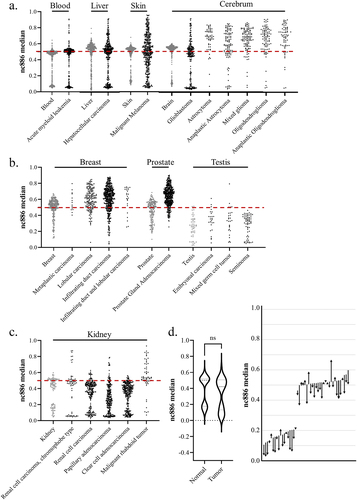
Similarly to Fort et al. [Citation37], we retrieved data from The Cancer Genome Atlas Program (TCGA) to investigate the nc886 methylation pattern in cancerous tissue, but unlike in previous literature, we investigated the methylation patterns instead of comparing mean methylation in the nc886 promoter region. In line with previous literature, we discovered that the changes in methylation are cancer-type-specific [Citation37]. While the methylation pattern in some cancers, such as acute myeloid leukaemia, remains mostly unaltered, in others, such as malignant melanoma, the bimodal methylation pattern observed in healthy skin is completely lost (). In the majority of cancer types, some degree of hypermethylation and loss of control of the methylation pattern can be observed (). In comparison to the bimodal pattern in blood, more systematic hypermethylation can be observed in brain cancers, with the exception of gliomas, as well as in prostate and breast cancers. Hypomethylation in comparison to healthy tissue can be observed in renal carcinomas. Both healthy and cancerous testes present lower methylation levels than blood, in addition to an absence of the bimodal methylation pattern. Similarly, in both healthy breast and prostate tissue, the bimodal methylation pattern is not present, as the majority of individuals present methylation levels of roughly 50%, and hypomethylation can be seen in tumours of these tissues ().
It will be interesting to study whether it is the methylation level itself, the degree of hypo- or hypermethylation, or the loss of the established pattern that is associated with cancerous development and future prognosis. One can hypothesize that the loss of imprint in this region could be a sign of epigenetic instability and thus associated with a poorer prognosis, regardless of the direction of change in the methylation levels. However, the original methylation pattern of the individual and tissues of origin, as well as the proportions of the methylation status groups in a population, should always be taken into account when interpreting the obtained results. For example, the findings from a cross-sectional study suggesting that the human papilloma virus is associated with similar changes in the nc886 methylation profile in both cancerous and healthy tissue could also be explained by different proportions of imprinted individuals in the case and control groups at baseline [Citation104]. Due to the peculiar nature of nc886 methylation, longitudinal analyses would be exceptionally beneficial to determine its role in cancer progression.
Conclusions
nc886 has been shown to be associated with both periconceptional conditions and adulthood health traits, but many of these results are not replicated in independent cohorts. Results obtained from small cohorts, in particular, warrant reanalysis and replication. Current knowledge indicates that the DNA methylation pattern in the nc866 locus is established during oocyte maturation [Citation6,Citation8], remains stable during life and in the majority of somatic tissues, but is associated with at least maternal age at birth [Citation1,Citation7]. In this case, it should be contemplated whether nc886 can be considered to mediate the DOHaD hypothesis, as the methylation status as such is not affected by environmental conditions. However, if the methylation pattern in a population is shaped by pregnancy success, nc886 could mediate the genetics-independent adaptation of the population to the surrounding conditions.
Approximately 75% of individuals of European ancestry present an imprinted nc886 locus, while populations of African descent present a higher percentage of imprinted individuals and Asian populations have systematically lower numbers of imprinted individuals. Genetic and epigenetic analyses of populations with more diverse backgrounds are needed to understand this pattern. In previous works, nc886 methylation has been associated with metabolic traits, and immortalization and carcinogenesis have been shown to alter the inherent DNA methylation pattern [Citation12]. We demonstrate herein that the changes in the methylation pattern are cancer-type- and tissue-of-origin-specific. However, the specific role of nc886 RNAs in the development of malignancy remains unclear. Similarly, the true nature of nc886 RNAs is still under debate. As the hairpin structure that has been suggested to bind PKR is formed by nucleotides 46–59 and the short RNAs are suggested to be processed from nucleotides 1–24 and 80–101, both hypotheses could potentially be correct.
Great inherent variation caused by the stable genetic and epigenetic regulators in the nc886 RNA expression within populations can lead to incorrect interpretations, when assuming that all differences between cases and controls are due to the studied condition. Furthermore, the binomial methylation pattern of nc886 warrants post-hoc analyses every time the region is discovered in an EWAS analysis. These notions can be generalized to study DNA methylation and the expression of gene products regulated via DNA methylation. Genetic variation and other features, such as sex, can contribute to methylation levels that are categorical rather than continuous variables. Especially in studies with small datasets, it would be important to inspect the distribution of identified methylation sites. Furthermore, even among continuous features, there is potentially physiological variation in the levels of the measured epigenetic profile that are not caused by the condition in question. Even though nc886 codes for only a few peculiar RNAs and is located in an atypical locus, the contradicting results presented herein highlight the fact that, in this era of genome-wide bioinformatic analyses and vast amounts of data, researchers should take time to study their top findings further in order to avoid reducing science to a mere reporting of statistically significant values.
Implications for future studies
The DNA methylation pattern in the nc886 locus should be treated as a binomial variable to avoid reporting false-positive findings due to the random distribution of non-methylated and imprinted individuals among cases and controls.
While studying the DNA methylation pattern in cancer, hypo- and hypermethylation of nc886 should be reported only when the intrinsic DNA methylation pattern has been taken into account in the analysis.
When investigating the associations between nc886 RNA levels and phenotypes, the stable genetic and epigenetic regulation pattern of the individuals or cell lines should be taken into account to avoid false-positive results caused by the uneven distribution of the stable regulatory profiles of nc886 RNAs in small case – control settings.
Open questions relating to nc886
How and when is the nc886 DMR established, and do the preconceptional/prenatal conditions modulate the DNA methylation pattern? Does the non-methylated methylation pattern, for example, provide a survival advantage to the foetus in non-optimal pregnancy conditions? What causes the distinct patterns of the nc886 methylation status in cohorts of different geographical origins? How are the intermediate and over-methylated nc886 methylation patterns established?
Does the functional form of nc886 RNAs constitute the 101nt-long hairpin structure binding to proteins, the two short RNAs produced by Dicer and acting in a miRNA-like manner, or both? What is the function of nc886 RNA(s) in physiological conditions?
Does nc886 have a causal role in carcinogenesis, or are the changes in DNA methylation pattern and RNA expression consequences of epigenetic instability?
Abbreviations
| CTCF | = | CCCTC-Binding Factor |
| DMR | = | differently methylated region |
| DOHaD | = | Developmental Origins of Health and Disease |
| EWAS | = | epigenome-wide association study |
| HDL | = | high-density lipoprotein |
| IgA | = | immunoglobulin A |
| iPSC | = | induced pluripotent stem cells |
| miR | = | microRNA |
| miRNA | = | microRNA |
| mQTL | = | methylation quantitative trait locus |
| nc886 | = | non-coding 886 |
| nt | = | nucleotide |
| OAS1 | = | 2–5-ologiadenylate synthetase 1 |
| PKR | = | protein kinase R |
| RNA pol III | = | RNA polymerase III |
| TCGA | = | The Cancer Genome Atlas |
| vtRNA2–1 | = | vault RNA2–1 |
Author contributions
ER conceptualized and wrote the first draft of the manuscript, provided data and figures, and reviewed and edited the manuscript. SR provided data and revised the manuscript. SM conceptualized, reviewed and edited the manuscript.
Ethical approval and consent to participate
All data utilized have been freely available, and the authors who have published the data are responsible for ethical approvals and consents to participate.
Odd duck_supplementary file.pdf
Download PDF (1.1 MB)Acknowledgments
We wish to thank Daria Kostiniuk (MSc) and Netta Pihlanen for their contribution to the manuscript. We would also like to thank all the researchers who have published on nc886 and especially those who have produced the data utilized herein and made it freely available or provided it upon request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2332819
Data availability statement
All new analyses performed have utilized data that are freely available or available upon reasonable request. For different geographic origins, data from Khomeni San (GSE99029 [Citation64], Gambian (GSE99863 [Citation118], Malawian and Jamaican (GSE112893 [Citation119], African American (GSE210255 [Citation120], Multiethnic American (GSE36369 [Citation65], Central African (https://ega-archive.org/EGAD00010000692; [Citation66], Gambian (GSE59592 [Citation13], Congolese (GSE224363 [Citation121], Latin American (GSE77716 [Citation122], individuals of European ancestry (summary statistics [Citation6], Bangladeshi (http://datadryad.org/10.5061/dryad.k67kf [Citation14], Chinese (GSE116379 [Citation78] Han Chinese (GSE201287 [Citation123], Taiwanese (GSE78904 [Citation124] and Indonesian (https://figshare.com,10.26188/5e00abd72f581, [Citation67] cohorts were utilized. In the reanalyses and attempts to replicate previous associations of nc886 methylation pattern and phenotypes, the following datasets were used: GSE52576 [Citation57] for parthenogenetically activated oocytes and embryonic stem cells; GSE59592 and GSE99863 for season of conception; http://datadryad.org/10.5061/dryad.k67kf, and GSE116379 for postnatal and prenatal exposure to famine; and GSE165081 [Citation81], GSE145361 [Citation83] and GSE111629 [Citation125] for the presence of Parkinson’s disease. Methylation data from tumours was downloaded from The Cancer Genome Atlas Program (TCGA), and the healthy reference tissues data were utilized as follows: brain (GSE72778 [Citation126], blood (GSE40279 [Citation127], liver (GSE61258 [Citation128] and GSE180474 [Citation129], skin (GSE90124 [Citation130], prostate (GSE76938 [Citation131] and GSE213478 [Citation132], breast (GSE88883 [Citation133], GSE101961 [Citation134] and GSE213478 [Citation132], testis (GSE213478 [Citation132] and kidney (GSE61441 [Citation135] and GSE213478 [Citation132].
Additional information
Funding
References
- Marttila S, Viiri LE, Mishra PP. et al. Methylation status of nc886 epiallele reflects periconceptional conditions and is associated with glucose metabolism through nc886 RNAs. Clin Epigenetics 2021;13(1). doi: 10.1186/s13148-021-01132-3
- Zink F, et al. Insights into imprinting from parent-of-origin phased methylomes and transcriptomes. Nature Genet. 2018;50(11):1542–20. doi: 10.1038/s41588-018-0232-7
- Dugué PA, Yu C, McKay T, et al. vtRNA2‐1: genetic variation, heritable methylation and disease association. Int J Mol Sci. 2021;22(5):1–18. doi: 10.3390/ijms22052535
- Dowty JG, et al. Heritable methylation marks associated with prostate cancer risk. Fam Cancer. 2023;22(3):313–317. doi: 10.1007/s10689-022-00325-w
- Dugué PA, et al. Heritable methylation marks associated with breast and prostate cancer risk. Prostate. 2018;78(13):962–969. doi: 10.1002/pros.23654
- Marttila S, et al. Methylation status of vtRNA2-1/nc886 is stable across human populations, monozygotic twin pairs and in majority of somatic tissues. 2022. doi: 10.1101/2022.06.21.496995
- Carpenter BL, et al. Mother–child transmission of epigenetic information by tunable polymorphic imprinting. Proc Natl Acad Sci, USA. 2018;115(51). doi: 10.1073/pnas.1815005115
- Carpenter BL, et al. Oocyte age and preconceptual alcohol use are highly correlated with epigenetic imprinting of a noncoding RNA (nc886). Proc Natl Acad Sci, USA. 2021;118(12). doi: 10.1073/pnas.2026580118
- Fort RS, Garat B, Sotelo-Silveira JR, et al. vtRNA2-1/nc886 produces a small RNA that contributes to its tumor suppression action through the microRNA pathway in prostate cancer. Noncoding RNA. 2020;6(1):7. doi: 10.3390/ncrna6010007
- Lee YS. Are we studying non-coding RNAs correctly? Lessons from nc886. Int J Mol Sci. 2022;23(8):4251. doi: 10.3390/ijms23084251
- Treppendahl MB, Qiu X, Søgaard A, et al. Allelic methylation levels of the noncoding vtRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood. 2012;119(1):206–216. doi: 10.1182/blood-2011-06-362541
- Park J-L, Lee Y-S, Song M-J, et al. Epigenetic regulation of RNA polymerase III transcription in early breast tumorigenesis. Oncogene. 2017;36(49):6793–6804. doi: 10.1038/onc.2017.285
- Silver MJ, et al. Independent genomewide screens identify the tumor suppressor vtRNA2-1 as a human epiallele responsive to periconceptional environment. Genome Bio. 2015;16(1). doi: 10.1186/s13059-015-0660-y
- Finer S, Iqbal MS, Lowe R, et al. Is famine exposure during developmental life in rural Bangladesh associated with a metabolic and epigenetic signature in young adulthood? A historical cohort study. BMJ Open. 2016;6(11):e011768. doi: 10.1136/bmjopen-2016-011768
- Lee Y-S, Lee YS. nc886, an RNA polymerase III-Transcribed noncoding RNA whose expression is dynamic and regulated by intriguing mechanisms. Int J Mol Sci. 2023;24(10):8533. doi: 10.3390/ijms24108533
- Minones-Moyano E, Friedländer MR, Pallares J, et al. Upregulation of a small vault RNA (vtRNA2-1a) is an early event in Parkinson disease and induces neuronal dysfunction. RNA Biol. 2013;10(7):1093–1106. doi: 10.4161/rna.24813
- Barker DJP, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in england and WALES. Lancet. 1986;327(8489):1077–1081. doi: 10.1016/S0140-6736(86)91340-1
- Landgraf P, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–1414. doi: 10.1016/j.cell.2007.04.040
- Yue J, Sheng Y, Orwig KE. Identification of novel homologous microRNA genes in the rhesus macaque genome. BMC Genomics. 2008;9(1):8. doi: 10.1186/1471-2164-9-8
- Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation. RNA. 2003;9(3):277–279. doi: 10.1261/rna.2183803
- Stadler PF, et al. Evolution of vault RNAs. Mol Biol Evol. 2009;26(9):1975–1991. doi: 10.1093/molbev/msp112
- Nandy C, Mrázek J, Stoiber H, et al. Epstein–barr virus-induced expression of a novel human vault RNA. J Mol Biol. 2009;388(4):776–784. doi: 10.1016/j.jmb.2009.03.031
- Mrázek J, Kreutmayer SB, Grässer FA, et al. Subtractive hybridization identifies novel differentially expressed ncRNA species in EBV-infected human B cells. Nucleic Acids Res. 2007;35(10):e73. doi: 10.1093/nar/gkm244
- Gallo S, Kong E, Ferro I, et al. Small but powerful: the human vault RNAs as multifaceted modulators of pro-survival characteristics and tumorigenesis. Cancers (Basel). 2022;14(11):2787. doi: 10.3390/cancers14112787
- Lee K, et al. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA. 2011;17(6):1076–1089. doi: 10.1261/rna.2701111
- Canella D, Praz V, Reina JH, et al. Defining the RNA polymerase III transcriptome: genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010;20(6):710–721. doi: 10.1101/gr.101337.109
- Kostiniuk D, Tamminen H, Mishra PP, et al. Methylation pattern of polymorphically imprinted nc886 is not conserved across mammalia. PloS One. 2022;17(3):e0261481. doi: 10.1371/journal.pone.0261481
- Kunkeaw N, Lee Y-S, Im WR, et al. Mechanism mediated by a noncoding RNA, nc886, in the cytotoxicity of a DNA-reactive compound. Proc Natl Acad Sci USA. 2019;116(17):8289–8294. doi: 10.1073/pnas.1814510116
- Ahn J-H, Lee H-S, Lee J-S, et al. nc886 is induced by TGF-β and suppresses the microRNA pathway in ovarian cancer. Nat Commun. 2018;9(1):1166. doi: 10.1038/s41467-018-03556-7
- Kunkeaw N, Jeon SH, Lee K, et al. Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene. 2013;32(32):3722–3731. doi: 10.1038/onc.2012.382
- Calderon BM, Conn GL. Human noncoding RNA 886 (nc886) adopts two structurally distinct conformers that are functionally opposing regulators of PKR. RNA. 2017;23(4):557–566. doi: 10.1261/rna.060269.116
- Calderon BM, Conn GL. A human cellular noncoding RNA activates the antiviral protein 2′–5′-oligoadenylate synthetase 1. J Biol Chem. 2018;293(41):16115–16124. doi: 10.1074/jbc.RA118.004747
- Lee YS. A novel type of non-coding RNA, nc886, implicated in tumor sensing and suppression. Genomics Inform. 2015;13(2):26. doi: 10.5808/gi.2015.13.2.26
- Sharbati J, et al. Integrated microRNA-mRNA-analysis of human monocyte derived macrophages upon mycobacterium avium subsp. hominissuis infection. PLoS One. 2011;6(5):e20258. doi: 10.1371/journal.pone.0020258
- Saruuldalai E, Park J, Kang D, et al. A host non-coding RNA, nc886, plays a pro-viral role by promoting virus trafficking to the nucleus. Mol Ther Oncolytics. 2022;24:683–694. doi: 10.1016/j.omto.2022.02.018
- Lee Y-S, Bao X, Lee H-H, et al. Nc886, a novel suppressor of the type I interferon response upon pathogen intrusion. Int J Mol Sci. 2021;22(4):2003. doi: 10.3390/ijms22042003
- Fort RS, Duhagon MA. Pan-cancer chromatin analysis of the human vtRNA genes uncovers their association with cancer Biology. 2021. doi: 10.12688/f1000research.28510.2
- Yu S, et al. Microarray analysis of differentially expressed microRnas in allergic rhinitis. Am J Rhinol Allergy. 2011. doi: 10.2500/ajra.2011.25.3682
- Suojalehto H, Lindström I, Majuri M-L, et al. Altered microRNA expression of nasal mucosa in long-term asthma and allergic rhinitis. Int Arch Allergy Immunol. 2014;163(3):168–178. doi: 10.1159/000358486
- Lee K-S, Cho E, Weon JB, et al. Inhibition of UVB-Induced inflammation by laminaria japonica extract via regulation of nc886-PKR pathway. Nutrients. 2020;12(7):1958. doi: 10.3390/nu12071958
- Lee K-S, Shin S, Cho E, et al. nc886, a non-coding RNA, inhibits UVB-induced MMP-9 and COX-2 expression via the PKR pathway in human keratinocytes. Biochem Biophys Res Commun. 2019;512(4):647–652. doi: 10.1016/j.bbrc.2019.01.068
- Joo JE, et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat Commun 2018;9(1). doi: 10.1038/s41467-018-03058-6
- Romanelli V, et al. Variable maternal methylation overlapping the nc886/vtRNA2-1 locus is locked between hypermethylated repeats and is frequently altered in cancer. Epigenetics. 2014;9(5):783–790. doi: 10.4161/epi.28323
- Hu Z, Zhang H, Tang L, et al. Silencing nc886, a non-coding RNA, induces apoptosis of human endometrial cancer cells-1A in vitro. Med Sci Monit. 2017;23:1317–1324. doi: 10.12659/MSM.900320
- Ross JP, van Dijk S, Phang M, et al. Batch-effect detection, correction and characterisation in Illumina HumanMethylation450 and MethylationEPIC BeadChip array data. Clin Epigenetics. 2022;14(1):58. doi: 10.1186/s13148-022-01277-9
- Lewis A, Reik W. How imprinting centres work. Cytogenet Genome Res. 2006;113(1–4):81–89. doi: 10.1159/000090818
- Pervjakova N, Kasela S, Morris AP, et al. Imprinted genes and imprinting control regions show predominant intermediate methylation in adult somatic tissues. Epigenomics. 2016;8(6):789–799. doi: 10.2217/epi.16.8
- Monk D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: the selected survival of imprints. Int J Biochem Cell Biol. 2015;67:128–138. doi: 10.1016/j.biocel.2015.04.014
- Baran Y, Subramaniam M, Biton A, et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015;25(7):927–936. doi: 10.1101/gr.192278.115
- Sanchez-Delgado M, Court F, Vidal E, et al. Human oocyte-derived methylation differences persist in the placenta revealing widespread transient imprinting. PloS Genet. 2016;12(11):e1006427. doi: 10.1371/journal.pgen.1006427
- Carli D, Riberi E, Ferrero GB, et al. Syndromic disorders caused by disturbed human imprinting. J Clin Res Pediatr Endocrinol. 2020;12(1):1–16. doi: 10.4274/jcrpe.galenos.2019.2018.0249
- Olsen KW, Castillo-Fernandez J, Zedeler A, et al. A distinctive epigenetic ageing profile in human granulosa cells. Hum Reprod. 2020;35(6):1332–1345. doi: 10.1093/humrep/deaa071
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230
- Lee H-S, Lee K, Jang H-J, et al. Epigenetic silencing of the non-coding RNA nc886 provokes oncogenes during human esophageal tumorigenesis. Oncotarget. 2014;5(11):3472–3481. doi: 10.18632/oncotarget.1927
- Lee K-S, Park J-L, Lee K, et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget. 2014;5(11):3944–3955. doi: 10.18632/oncotarget.2047
- Sallustio F, Serino G, Cox S, et al. Aberrantly methylated DNA regions lead to low activation of CD4+ T-cells in IgA nephropathy. Clin Sci. 2016;130(9):733–746. doi: 10.1042/CS20150711
- Court F, Tayama C, Romanelli V, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24(4):554–569. doi: 10.1101/gr.164913.113
- Okae H, Chiba H, Hiura H, et al. Genome-wide analysis of DNA methylation dynamics during early human development. PloS Genet. 2014;10(12):e1004868. doi: 10.1371/journal.pgen.1004868
- Jima DD, Skaar DA, Planchart A, et al. Genomic map of candidate human imprint control regions: the imprintome. Epigenetics. 2022;17(13):1920–1943. doi: 10.1080/15592294.2022.2091815
- Zhu P, Guo H, Ren Y, et al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet. 2018;50(1):12–19. doi: 10.1038/s41588-017-0007-6
- Obata Y, Kono T. Maternal primary imprinting is established at a specific time for each gene throughout oocyte growth*. J Biol Chem. 2002;277(7):5285–5289. doi: 10.1074/jbc.M108586200
- Guo H, Zhu P, Yan L, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511(7511):606–610. doi: 10.1038/nature13544
- You Y-A, Kwon EJ, Hwang H-S, et al. Elevated methylation of the vault RNA2-1 promoter in maternal blood is associated with preterm birth. BMC Genomics. 2021;22(1):528. doi: 10.1186/s12864-021-07865-y
- Gopalan S, Carja O, Fagny M, et al. Trends in DNA methylation with age replicate across diverse human populations. Genetics. 2017;206(3):1659–1674. doi: 10.1534/genetics.116.195594
- Heyn H, Moran S, Hernando-Herraez I, et al. DNA methylation contributes to natural human variation. Genome Res. 2013;23(9):1363–1372. doi: 10.1101/gr.154187.112
- Fagny M, Patin E, MacIsaac JL, et al. The epigenomic landscape of African rainforest hunter-gatherers and farmers. Nat Commun. 2015;6(1):10047. doi: 10.1038/ncomms10047
- Natri HM, Bobowik KS, Kusuma P, et al. Genome-wide DNA methylation and gene expression patterns reflect genetic ancestry and environmental differences across the Indonesian archipelago. PLoS Genet. 2020;16(5):e1008749. doi: 10.1371/journal.pgen.1008749
- Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177(1):26–31. doi: 10.1016/j.cell.2019.02.048
- Markunas CA, et al. Maternal age at delivery is associated with an epigenetic signature in both newborns and adults. PLoS One. 2016;11(7):e0156361. doi: 10.1371/journal.pone.0156361
- Richmond RC, Sharp GC, Herbert G, et al. The long-term impact of folic acid in pregnancy on offspring DNA methylation: follow-up of the aberdeen folic acid supplementation trial (AFAST). Int J Epidemiol. 2018;47(3):928–937. doi: 10.1093/ije/dyy032
- Van Der Plaat DA, et al. Occupational exposure to pesticides is associated with differential DNA methylation. Occup Environ Med. 2018;75(6):427–435. doi: 10.1136/oemed-2017-104787
- van der Plaat DA, Vonk JM, Terzikhan N, et al. Occupational exposure to gases/fumes and mineral dust affect DNA methylation levels of genes regulating expression. Hum Mol Genet. 2019;28(15):2477–2485. doi: 10.1093/hmg/ddz067
- Ambatipudi S, Cuenin C, Hernandez-Vargas H, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8(5):599–618. doi: 10.2217/epi-2016-0001
- Xu R, Li S, Wu Y, et al. Wildfire-related PM2.5 and DNA methylation: an Australian twin and family study. Environ Int. 2023;171:107704. doi: 10.1016/j.envint.2022.107704
- Eze IC, Jeong A, Schaffner E, et al. Genome-wide DNA methylation in peripheral blood and long-term exposure to source-specific transportation noise and air pollution: the SAPALDIA study. Environ Health Perspect. 2020;128(6):067003. doi: 10.1289/EHP6174
- Tobi EW, Goeman JJ, Monajemi R, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5(1):5592. doi: 10.1038/ncomms6592
- Peter CJ, Fischer LK, Kundakovic M, et al. DNA methylation signatures of early childhood malnutrition associated with impairments in attention and cognition. Biological Psychiatry. 2016;80(10):765–774. doi: 10.1016/j.biopsych.2016.03.2100
- Boks MP, Houtepen LC, Xu Z, et al. Genetic vulnerability to DUSP22 promoter hypermethylation is involved in the relation between in utero famine exposure and schizophrenia. NPJ Schizophr. 2018;4(1):1–8. doi: 10.1038/s41537-018-0058-4
- Van Dijk SJ, Peters TJ, Buckley M, et al. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int J Obesity. 2018;42(1):28–35. doi: 10.1038/ijo.2017.228
- Gonseth S, Shaw GM, Roy R, et al. Epigenomic profiling of newborns with isolated orofacial clefts reveals widespread DNA methylation changes and implicates metastable epiallele regions in disease risk. Epigenetics. 2019;14(2):198–213. doi: 10.1080/15592294.2019.1581591
- Henderson AR, Wang Q, Meechoovet B, et al. DNA methylation and expression profiles of whole blood in Parkinson’s disease. Front Genet. 2021;12. doi: 10.3389/fgene.2021.640266
- Chuang YH, Paul KC, Bronstein JM, et al. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017;9(1). doi: 10.1186/s13073-017-0466-5
- Vallerga CL, Zhang F, Fowdar J, et al. Analysis of DNA methylation associates the cystine–glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat Commun. 2020;11(1):1238. doi: 10.1038/s41467-020-15065-7
- Sallustio F, Picerno A, Cimmarusti MT, et al. Elevated levels of IL-6 in IgA nephropathy patients are induced by an epigenetically driven mechanism modulated by viral and bacterial RNA. Eur J Internal Med. 2023;118:108–117. doi: 10.1016/j.ejim.2023.07.045
- Li JH, Xiao X, Zhang Y-N, et al. MicroRNA miR-886-5p inhibits apoptosis by down-regulating Bax expression in human cervical carcinoma cells. Gynecol Oncol. 2011;120(1):145–151. doi: 10.1016/j.ygyno.2010.09.009
- Kong L, Hao Q, Wang Y, et al. Regulation of p53 expression and apoptosis by vault RNA2-1-5p in cervical cancer cells. Oncotarget. 2015;6(29):28371–28388. doi: 10.18632/oncotarget.4948
- Li J-H, Wang M, Zhang R, et al. E2F1-directed activation of nc886 mediates drug resistance in cervical cancer cells via regulation of major vault protein. Int J Clin Exp Pathol. 2017;10(9):9233–9242.
- Zhang L-L, Wu J, Liu Q, et al. MiR-886-5p inhibition inhibits growth and induces apoptosis of MCF7 cells. Asian Pac J Cancer Prev. 2014;15(4):1511–1515. doi: 10.7314/APJCP.2014.15.4.1511
- Khoshnevisan A, Parvin M, Ghorbanmehr N, et al. A significant upregulation of miR-886-5p in high grade and invasive bladder tumors. Urol J. 2015;12(3):2160–2164.
- McDonald AC, Vira M, Walter V, et al. Circulating microRnas in plasma among men with low-grade and high-grade prostate cancer at prostate biopsy. Prostate. 2019;79(9):961–968. doi: 10.1002/pros.23803
- Pan B, Yu J, Liu X. Upregulation of miR-886 indicates poor prognosis and promotes tumour progression of prostate cancer. Andrologia. 2022;54(1):e14296. doi: 10.1111/and.14296
- Xiang P, Liu Y, Liu L, et al. The biological function and clinical significance of miR-886-5p in multiple myeloma. AHA. 2019;142(4):208–216. doi: 10.1159/000499620
- Lei J, Xiao J-H, Zhang S-H, et al. Non-coding RNA 886 promotes renal cell carcinoma growth and metastasis through the janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Mol Med Rep. 2017;16(4):4273–4278. doi: 10.3892/mmr.2017.7093
- Jang H-J, Lee H-S, Burt BM, et al. Integrated genomic analysis of recurrence-associated small non-coding RNAs in oesophageal cancer. Gut. 2017;66(2):215–225. doi: 10.1136/gutjnl-2015-311238
- Fort RS, Mathó C, Geraldo MV, et al. Nc886 is epigenetically repressed in prostate cancer and acts as a tumor suppressor through the inhibition of cell growth. BMC Cancer 2018;18(1). doi: 10.1186/s12885-018-4049-7
- Yang R, Zuo L, Ma H, et al. Downregulation of nc886 contributes to prostate cancer cell invasion and TGFβ1-induced EMT. Genes Dis. 2022;9(4):1086–1098. doi: 10.1016/j.gendis.2020.12.010
- Liang S, et al. Screening and identification of potential miRNA involved in ovarian cancer invasion and metastasis. Zhonghua Zhong Liu Za Zhi. 2010;32(9):650–654.
- Tahiri A, Leivonen S-K, Lüders T, et al. Deregulation of cancer-related miRnas is a common event in both benign and malignant human breast tumors. Carcinogenesis. 2014;35(1):76–85. doi: 10.1093/carcin/bgt333
- Xiong Y, et al. MiR-886-3p regulates cell proliferation and migration, and is dysregulated in familial non-medullary thyroid cancer. PLoS One. 2011;6(10):e24717. doi: 10.1371/journal.pone.0024717
- Dettmer MS, et al. MicroRNA profile of poorly differentiated thyroid carcinomas – new diagnostic and prognostic insights. J Mol Endocrinol. 2014;52(2):181–189. doi: 10.1530/JME-13-0266
- Bi N, Cao J, Song Y, et al. A microRNA signature predicts survival in early stage small-cell lung cancer treated with surgery and adjuvant chemotherapy. PLoS One. 2014;9(3):e91388. doi: 10.1371/journal.pone.0091388
- Gao W, Shen H, Liu L, et al. MiR-21 overexpression in human primary squamous cell lung carcinoma is associated with poor patient prognosis. J Cancer Res Clin Oncol. 2011;137(4):557–566. doi: 10.1007/s00432-010-0918-4
- Zhang Y, Wang X, Han S, et al. Suppression of mir-886-3P mediated by Arecoline (are) contributes to the progression of oral squamous cell carcinoma. J Invest Med. 2021;69(2):377–381. doi: 10.1136/jim-2020-001405
- Xu Y, Wang Z, Wei P, et al. Hypermethylation of nc886 in HPV-positive oropharyngeal cancer and its clinical implications: an epigenome-wide association study. Mol Ther Nucleic Acids. 2022;30:596–605. doi: 10.1016/j.omtn.2022.11.012
- Im WR, Lee H-S, Lee Y-S, et al. A regulatory noncoding RNA, nc886, suppresses esophageal cancer by inhibiting the AKT pathway and cell cycle progression. Cells. 2020;9(4):801. doi: 10.3390/cells9040801
- Gao W, Zhang S, Guorong L, et al. Nc886 promotes renal cancer cell drug-resistance by enhancing EMT through Rock2 phosphorylation-mediated β-catenin nuclear translocation. Cell Cycle. 2022;21(4):340–351. doi: 10.1080/15384101.2021.2020431
- Nordentoft I, Birkenkamp-Demtroder K, Agerbæk M, et al. miRnas associated with chemo-sensitivity in cell lines and in advanced bladder cancer. BMC Med Genomics. 2012;5(1):40. doi: 10.1186/1755-8794-5-40
- Cao J, et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013;73(11):3326–3335. doi: 10.1158/0008-5472.CAN-12-3055
- Shen J, Zhou W, Bi N, et al. MicroRNA-886-3P functions as a tumor suppressor in small cell lung cancer. Cancer Biol Ther. 2018;19(12):1185–1192. doi: 10.1080/15384047.2018.1491505
- Schou JV, Rossi S, Jensen BV, et al. miR-345 in metastatic colorectal cancer: a non-invasive biomarker for clinical outcome in non-KRAS mutant patients treated with 3rd line cetuximab and Irinotecan. PLoS One. 2014;9(6):e99886. doi: 10.1371/journal.pone.0099886
- Okumura T, Kojima H, Miwa T, et al. The expression of microRNA 574-3p as a predictor of postoperative outcome in patients with esophageal squamous cell carcinoma. World J Surg Oncol. 2016;14(1):228. doi: 10.1186/s12957-016-0985-3
- Lee EK, et al. Nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget. 2016;7(46):75000–75012. doi: 10.18632/oncotarget.11852
- Ortega-García MB, et al. Uncovering tumour heterogeneity through PKR and nc886 analysis in metastatic colon cancer patients treated with 5-FU-based chemotherapy. Cancers (Basel). 2020;12(2):379. doi: 10.3390/cancers12020379
- Han Z-B, Zhong L, Teng M-J, et al. Identification of recurrence-related microRnas in hepatocellular carcinoma following liver transplantation. Mol Oncol. 2012;6(4):445–457. doi: 10.1016/j.molonc.2012.04.001
- Yu M-C, Lee C-W, Lin C-H, et al. Differential hypermethylation of the vtRNA2-1 promoter in hepatocellular carcinoma as a prognostic factor: tumor marker prognostic study. Int J Surg. 2020;79:282–289. doi: 10.1016/j.ijsu.2020.05.016
- Wang Z, Lu Y, Fornage M, et al. Identification of novel susceptibility methylation loci for pancreatic cancer in a two-phase epigenome-wide association study. Epigenetics. 2022;17(11):1357–1372. doi: 10.1080/15592294.2022.2026591
- Paliwal A, et al. Comparative anatomy of chromosomal domains with imprinted and non-imprinted allele-specific DNA methylation. PLoS Genet. 2013;9(8):e1003622. doi: 10.1371/journal.pgen.1003622
- Van Baak TE, et al. Epigenetic supersimilarity of monozygotic twin pairs. Genome Bio. 2018;19(2). doi: 10.1186/s13059-017-1374-0
- Schulze KV, Swaminathan S, Howell S, et al. Edematous severe acute malnutrition is characterized by hypomethylation of DNA. Nat Commun. 2019;10(1):5791. doi: 10.1038/s41467-019-13433-6
- Kho M, et al. Epigenetic loci for blood pressure are associated with hypertensive target organ damage in older African Americans from the genetic epidemiology network of arteriopathy (GENOA) study. BMC Med Genomics. 2020;13(1):131. doi: 10.1186/s12920-020-00791-0
- Quinn EB, Hsiao CJ, Maisha FM, et al. Prenatal maternal stress is associated with site-specific and age acceleration changes in maternal and newborn DNA methylation. Epigenetics. 2023;18(1):2222473. doi: 10.1080/15592294.2023.2222473
- Galanter JM, Gignoux CR, Oh SS, et al. Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures. Elife. 2017;6:e20532. doi: 10.7554/eLife.20532
- Xiu J, Li J, Liu Z, et al. Elevated BICD2 DNA methylation in blood of major depressive disorder patients and reduction of depressive-like behaviors in hippocampal Bicd2-knockdown mice. Proc Natl Acad Sci U S A. 2022;119(30):e2201967119. doi: 10.1073/pnas.2201967119
- Kao W-Y, Yang S-H, Liu W-J, et al. Genome-wide identification of blood DNA methylation patterns associated with early-onset hepatocellular carcinoma development in hepatitis B carriers. Mol Carcinog. 2017;56(2):425–435. doi: 10.1002/mc.22505
- Horvath S, Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015;7:1130–1142. doi:10.18632/aging.100859
- Horvath S, et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY). 2016;8:1485–1512. doi: 10.18632/aging.101005
- Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016
- Horvath S, Erhart W, Brosch M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci USA. 2014;111(43):15538–15543. doi: 10.1073/pnas.1412759111
- Johnson ND, Wu X, Still CD, et al. Differential DNA methylation and changing cell-type proportions as fibrotic stage progresses in NAFLD. Clin Epigenetics. 2021;13(1):152. doi: 10.1186/s13148-021-01129-y
- Roos L, Sandling JK, Bell CG, et al. Higher nevus Count exhibits a distinct DNA methylation signature in healthy human skin: implications for melanoma. J Invest Dermatol. 2017;137(4):910–920. doi: 10.1016/j.jid.2016.11.029
- Kirby MK, Ramaker RC, Roberts BS, et al. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer. 2017;17(1):273. doi: 10.1186/s12885-017-3252-2
- Oliva M, Demanelis K, Lu Y, et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat Genet. 2023;55(1):112–122. doi: 10.1038/s41588-022-01248-z
- Johnson KC, Houseman EA, King JE, et al. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. 2017;19(1):81. doi: 10.1186/s13058-017-0873-y
- Song M-A, Brasky TM, Weng DY, et al. Landscape of genome-wide age-related DNA methylation in breast tissue. Oncotarget. 2017;8(70):114648–114662. doi: 10.18632/oncotarget.22754
- Wei J-H, Haddad A, Wu K-J, et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat Commun. 2015;6(1):8699. doi: 10.1038/ncomms9699