Abstract
A reasonably efficient two-step synthesis of N-(2-ethylhexyl)-2,7-diiodocarbazole is presented. In the first step, the 4,4′-diiodobiphenyl was nitrated to a mixture of 4,4′-diiodo-2-nitrobiphenyl and 4-iodo-4′-nitrobiphenyl. In the second step, the mixture of these compounds was converted by simultaneous carbazole ring closure and N-alkylation to the N-(2-ethylhexyl)-2,7-diiodocarbazole by means of tris(2-ethylhexyl) phosphite. The synthesis represents a shorter alternative to the known more tedious procedures. The prepared compound is a suitable monomer for synthesis of conjugated polymers for optoelectronic applications.
1. Introduction
Poly(2,7-carbazole)s as an example of conjugated polymers have drawn much attention over the past decades due to their great potential for numerous electrical and optical applications Citation[1–4], such as organic light-emitting diodes, solar cells, and field-effect transistors. The synthesis of carbazole containing conjugated polymers and copolymers demands, besides others, 2,7-dihalocarbazole monomers. Whereas 3,6-functionalized carbazoles can be readily synthesized by electrophilic aromatic substitution, the preparation of 2,7-substituted structures is not as straightforward. Usually, the 4,4′-dihalo-2-nitrobiphenyl precursor units and Cadogan ring closure Citation[5] were utilized for syntheses of 2,7-chlorine or bromine substituted carbazole derivatives. As follows, efficient (three-step) procedures were developed for the preparation of N-(2-ethylhexyl)-2,7-dichlorocarbazole Citation[6] and N-(2-ethylhexyl)-2,7-dibromocarbazole Citation[7]. However, in the case of the diiodo derivative, N-(2-ethylhexyl)-2,7-diiodocarbazole (1), a more complicated five-step synthesis based on an azide strategy was reported Citation[6,8]. In the procedure, first a commercially available 2-amino-4,4′-dinitrobiphenyl is converted via the diazonium salt (1. NaNO2 and 2. NaN3) to 2-azido-4,4′-dinitrobiphenyl. The thermal carbazole ring closure gives the 2,7-dinitrocarbazole in the second step. This compound was reduced (SnCl2) to 2,7-diaminocarbazole (step 3) and, again via the diazonium salt (1. NaNO2 and 2. KI), was converted to 2,7-diidocarbazole (step 4). Finally, the N-(2-ethylhexyl)-2,7-diiodocarbazole was obtained by N-alkylation using 2-ethylhexyl bromide (NaOH) in the fifth step. The overall yields were reported as 14 and 17% (Citation[6],Citation[8], respectively). Recently, we published a shorter three-step synthesis of 1, where an overall yield of 18% was reached Citation[9]. The synthesis represented a significant simplification of the five-step procedure mentioned above. The commercially available 4,4′-diiodobiphenyl (2) was nitrated and the resulting 4,4′-diiodo-2-nitrobiphenyl (3) was converted via Freeman’s modification Citation[10] of the Cadogan ring closure to 2,7-diiodocarbazole (4) which was then N-alkylated in the final step to 1 (Scheme ).
Scheme 1 Three-step synthesis of N-(2-ethylhexyl)-2,7-diiodocarbazole (1) (overall yield: 18% Citation[9]).
![Scheme 1 Three-step synthesis of N-(2-ethylhexyl)-2,7-diiodocarbazole (1) (overall yield: 18% Citation[9]).](/cms/asset/40217a08-1c9b-4c7c-b277-3c8949e60d1b/tdmp_a_705487_o_f0006g.gif)
Here, we report a new two-step synthesis of N-(2-ethylhexyl)-2,7-diiodocarbazole (1), which represents further simplification of the mentioned three-step procedure and, simultaneously, we indicate other possibilities in the syntheses of N-alkyl-2,7-dihalocarbazoles. Generally, these materials are important in the preparation of the corresponding diboronic acid esters for Suzuki coupling Citation[11,12], which belongs to the array of important synthetic methods for preparation of conjugated polymers. The reactive diiodo derivatives are important in the preparation of such esters [e.g. N-(2-ethylhexyl)-2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)carbazole], because they sometimes give better yields in both the diboronic acid ester synthesis and Suzuki coupling. The first step in the synthesis of aromatic diboronic acid esters is a transmetallation reaction of the aryldihalogenide (X–Ar–X) with an organometallic compound (R–Li),
which can be complicated with a side reaction
The side reaction is significantly less important for X = I than for X = Br, since the iodine–lithium exchange is several orders of magnitude faster than the bromine–lithium exchange. The utilization of iodine compounds in the syntheses was limited because of their more complicated preparation in comparison with bromo or chloro derivatives. Our new short synthesis of diodo derivative gives the possibility to overcome this disadvantage in the case of the carbazole unit mentioned here.
2 Experimental
2.1 Materials and general procedures
The starting 4,4′-diiodobiphenyl, tris(2-ethylhexyl) phosphite, and the other chemicals were purchased from Aldrich or TCI and used directly without purification. Silicagel 60 (0.063–0.200 mm, Merck) was used for column chromatography (columns Ø 3–4 × 60 cm). TLC was performed with Silicagel 60 F254 aluminum sheets (Merck) and the R F values have their common meaning (R F = distance traveled by substance/distance traveled by solvent front). 1H and 13C NMR spectra were measured in CDCl3 or deuterated dimethylsulfoxide with an upgraded Bruker Advance DPX-300 spectrometer at 300.13 and 75.45 MHz, respectively, with hexamethyldisiloxane as internal standard. FT IR spectra were measured on a Perkin-Elmer Paragon 1000 PC Fourier transform infrared spectrometer by means of diamond ATR. Gas chromatography-mass spectrometry (GC-MS) measurements were performed with a Perkin Elmer gas chromatograph Auto System XL equipped with mass spectrometer Turbo Mass.
2.2 Synthesis and characterization
2.2.1 4,4′-Diiodo-2-nitrobiphenyl (3) and 4-iodo-4′-nitrobiphenyl (5)
The 4,4′-diiodobiphenyl (2, 6.09 g, 0.015 mol) was heated to 40 °C in acetic acid (80 mL), where it was not completely dissolved. Fuming (100%) nitric acid (25 mL) was slowly added (ca. 5 min) under stirring. The temperature was increased to 80 °C and kept for 30 min (everything was dissolved) and then it was left to cool to room temperature. Water (250 mL) was added and a yellow precipitate was filtered off (S2), washed with water and dried (yield: 5.50 g). This raw material was crystallized from ethanol and yellow purified material was obtained (yield: 4.25 g). This mixture of compounds (according to GC-MS mainly 3 + 5) was chromatographed on silicagel in toluene/heptane (1:1 by vol.). The separated corresponding fractions of 3 (R F = 0.68) and 5 (R F = 0.46) were combined and evaporated. The chromatographically pure compounds 3 (yield: 2.15 g (32%), mp 154–155 °C, lit Citation[13] 156 °C) and 5 (yield: 1.86 g (38%), mp 214–215 °C, lit Citation[14] 212–214 °C) were obtained.
2.2.1.1 Analysis of 3
Anal. calcd for C12H7I2NO2 (451.0) (3): C, 31.96; H, 1.56; N, 3.11; and I, 56.28. Found: C, 32.16; H, 1.50; N, 3.22; and I, 56.11.
1H NMR (300.1 MHz, CDCl3/DMSO-d 6 = 3:1 by vol.) (3): δ = 8.29 (d, J = 8.1 Hz, 1H, aromatic), 8.08 (d, J = 8.1 Hz, 1H, aromatic), 7.80 (d, 2H, J = 7.8 Hz, aromatic), 7.29 (d, J = 8.1 Hz, 1H, aromatic), and 7.11 (d, J = 8.1 Hz, 2H, aromatic).
13C NMR (75 MHz, CDCl3/DMSO-d 6 = 3:1 by vol.) (3): δ = 93.8 (1C), 95.1 (1C), 129.7 (2C), 132.2 (1C), 133.2 (1C), 133.6 (1C), 135.8 (1C), 137.6 (2C), 144.2 (1C), and 149.0 (1C), all aromatic.
2.2.1.2 Analysis of 5
Anal. calcd for C12H8INO2 (325.1) (5): C, 44.33; H, 2.48; N, 4.31; and I, 39.04. Found: C, 44.20; H, 2.47; N, 4.27; and I, 39.16.
1H NMR (300.1 MHz, CDCl3/DMSO-d 6 = 3:1 by vol.) (5): δ = 8.29 (d, J = 9.0 Hz, 2H, aromatic), 7.92 (d, J = 9.0 Hz, 2H, aromatic), 7.87 (d, J = 10.8 Hz, 2H, aromatic), and 7.55 (d, J = 8.7 Hz, 2H, aromatic).
13C NMR (75 MHz, CDCl3/DMSO-d 6 = 3:1 by vol.) (5): δ = 95.8 (1C), 124.1 (2C), 127.7 (2C), 129.3 (2C), 137.4 (1C), 138.0 (2C), 145.7 (1C), and 146.9 (1C), all aromatic.
2.2.2 N-(2-Ethylhexyl)-2,7-diiodocarbazole (1)
4,4′-Diiodo-2-nitrobiphenyl (3, 1.804 g, 0.004 mol), 4-iodo-4′-nitrobiphenyl (5, 1.951 g, 0.006 mol), and tris(2-ethylhexyl) phosphite (20.93 g, 0.05 mol) were heated at 160 °C under argon for 10 h. After cooling, the reaction mixture was chromatographed on silica gel in toluene/heptane (1:1 by vol.). The nicely separated fractions of the major product 1 (R F = 0.90) and minor product 4 (R F = 0.56) were combined and evaporated. The chromatographically pure compounds 1 (yield: 0.85 g (40%), mp 110–111 °C, lit Citation[9] mp 110–111 °C) and 4 (yield: 0.25 g (15%), mp 284–285 °C, lit Citation[15] mp 265–266 °C) were obtained. Products 1 and 4 were recrystallized from methanol for melting point, FT IR, 1H and 13C NMR measurements.
2.2.2.1 Analysis of 1
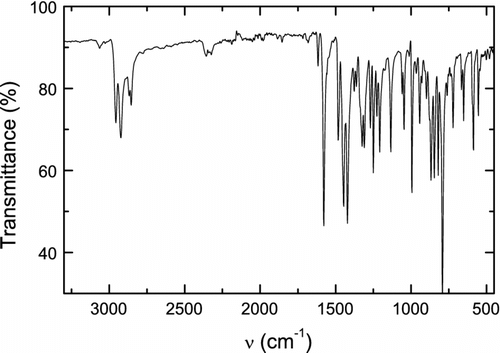
FT IR (ATR, Figure ): 3063 (aromatic C–H stretch), 2956, 2922, 2868, and 2854 (aliphatic C–H stretch), 1855, 1716, and 1682 (overtone bands), 1616, 1578, 1483, 1446, and 1421 (ring stretch), 1324 and 1309 (N-ring stretch), 1134 (C–N stretch), 1269, 1249, 1046, 994, and 942 (C–H in-plane), 867, 846, 820, and 792 (C–H out-of-plane), 586 (C–I stretch), and 554, 501, and 480 cm−1 (ring deformation).
Figure 1 FT IR (ATR) spectrum of N-(2-ethylhexyl)-2,7-diiodocarbazole (1).
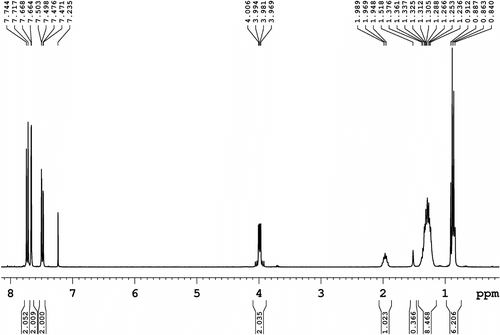
1H NMR (300.1 MHz, CDCl3, Figure ): δ = 7.73 (d, 2H, aromatic, J = 8.1 Hz), 7.66 (s, 2H, aromatic), 7.48 (d, 2H, aromatic, J = 6.6 Hz), 3.99 (d, 2H, N–CH2, J = 11.1 Hz), 1.99–1.94 (m, 1H, =CH–), 1.37–1.23 (m, 8H, 4 × CH2), and 0.91–0.84 (m, 6H, 2 × CH3).
Figure 2 1H NMR spectrum (300.1 MHz, CDCl3) of N-(2-ethylhexyl)-2,7-diiodocarbazole (1).
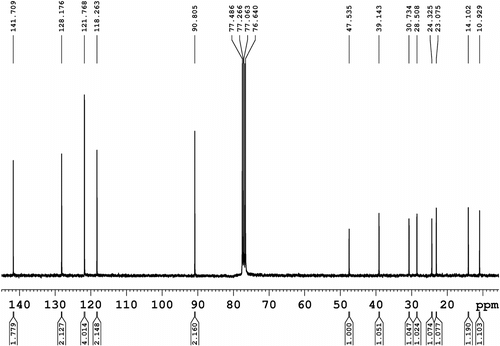
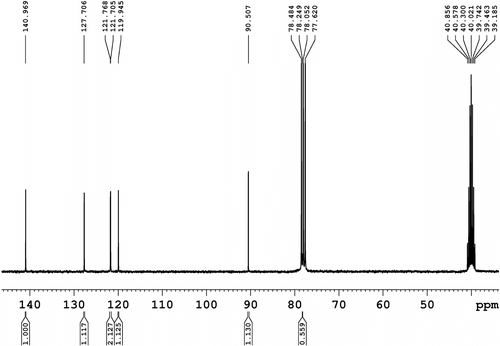
13C NMR (75 MHz, CDCl3, Figure ): δ = 10.9, 14.1, 23.1, 24.3, 28.5, 30.7, 39.1, and 47.5 (8C, aliphatic) and 90.8 (2C), 118.3 (2C), 121.8 (4C), 128.2 (2C), and 141.7 (2C), all aromatic.
Figure 3 13C NMR spectrum (75 MHz, CDCl3) of N-(2-ethylhexyl)-2,7-diiodocarbazole (1).
Anal. calcd for C20H23NI2 (531.2): C, 45.22; H, 4.36; N, 2.64; and I, 47.78. Found: C, 45.29; H, 4.21; N, 2.61; and I, 47.84.
Analysis of 4 : It was in agreement with the data shown below.
2.2.3 2,7-Diiodocarbazole (4)
4,4′-Diiodo-2-nitrobiphenyl (3, 1.804 g, 0.004 mol) and tris(2-ethylhexyl) phosphite (20.93 g, 0.05 mol) were heated at 160 °C under argon for 10 h. After cooling, the reaction mixture was chromatographed on silica gel in toluene/heptane (1:1 by vol.). The separated fractions of the major product 4 (R F = 0.56) and minor product 1 (R F = 0.90) were combined and evaporated. The chromatographically pure compounds 4 (yield: 0.97 g (58%)) and 1 (yield: 0.06 g (3%)) were obtained.
2.2.3.1 Analysis of 4
FT IR (ATR): 3389 (N–H stretch), 3067 (C–H stretch), 1980, 1888, and 1714 (overtone bands), 1590, 1471, 1453, and 1416 (ring stretch), 1135 (C–N stretch), 1237, 1043, 993, and 942 (C–H in-plane), 858, 819, and 800 (C–H out-of-plane), 727 (N–H wag), 523 (C–I stretch), and 499 cm−1 (C–N–C bend).
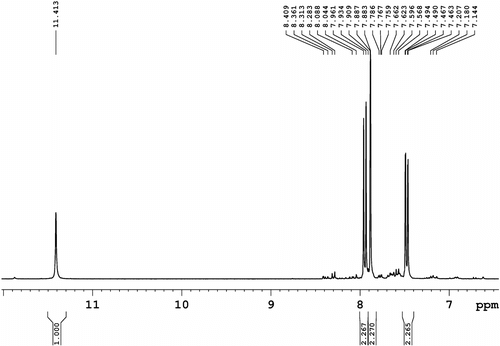
1H NMR (300.1 MHz, CDCl3/DMSO-d 6, 3:1, v/v, Figure ): δ = 11.41 (s, 1H, N–H), 7.95 (d, J = 8.1 Hz, 2H, aromatic), 7.88 (s, 2H, aromatic), and 7.48 (d, J = 9.0 Hz, 2H, aromatic).
Figure 4 1H NMR spectrum (300.1 MHz, CDCl3/DMSO-d 6, 3:1, v/v) of 2,7-diiodocarbazole (4).
13C NMR (75 MHz, CDCl3/DMSO-d 6, 3:1, v/v, Figure ): δ = 90.5 (2C), 120.0 (2C), 121.7 (2C), 121.8 (2C), 127.7 (2C), and 141.0 (2C), (all aromatic).
Figure 5 13C NMR spectrum (75 MHz, CDCl3/DMSO-d 6, 3:1, v/v) of 2,7-diiodocarbazole (4).
Anal. calcd for C12H7NI2 (419.0): C, 34.40; H, 1.68; N, 3.34; and I, 60.57. Found: C, 34.66; H, 1.57; N, 3.32; and I, 60.26.
Analysis of 1 was in agreement with the data shown above.
3 Results and discussion
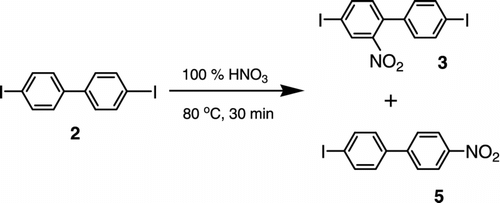
The first step of the N-(2-ethylhexyl)-2,7-diiodocarbazole (1) synthesis is the nitration of commercial 4,4′-diiodobiphenyl (2) according to Scheme . We tuned the nitration reaction to obtain a product composed of the 4,4′-diiodo-2-nitrobiphenyl (3) and 4-iodo-4′-nitrobiphenyl (5) in a weight ratio of approximately 1:1. The nitration of 2 was performed with 100% HNO3 in acetic acid at 80 °C for 30 min. After cooling, the nitration causes precipitation of raw material, which was crystallized from ethanol to give a purified yellow mixture of compounds. GC-MS analysis of this mixture revealed the presence of compounds 3 (50 wt.%), 5 (45 wt.%), 4-iodobiphenyl (2 wt.%), and 2 (1 wt.%) (contents of compounds < 1 wt.% were neglected). Pure compounds 3 and 5 were isolated by a column chromatography on silica gel in toluene/heptane (1:1 by vol.) and characterized (1H and 13C NMR, elemental analysis, melting point, TLC). The R F values determined by TLC in toluene/heptane (1:1 by vol.) were 0.68 and 0.46 for 3 and 5, respectively.
Scheme 2 The nitration of 4,4′-diiodobiphenyl (2) to 4,4′-diiodo-2-nitrobiphenyl (3) and 4-iodo-4′-nitrobiphenyl (5) – step 1.
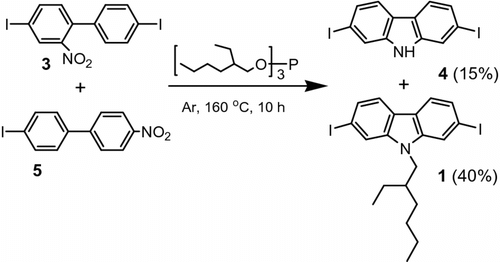
In the second step, the carbazole ring closure and N-alkylation was accomplished using tris(2-ethylhexyl) phosphite in the reaction mixture consisting of compounds 3 and 5. The reaction of 3 and 5 (1:1.5 by mol) with an excess of tris(2-ethylhexyl) phosphite in argon at 160 °C proceeded smoothly giving rise the N-(2-ethylhexyl)-2,7-diiodocarbazole (1) as the major product (40%) and 4 as a minor product (15%) (Scheme ).
Scheme 3 Tris(2-ethylhexyl) phosphite mediated carbazole ring closure applied on compounds 3 + 5 gives N-(2-ethylhexyl)-2,7-diiodocarbazole (1) as a major product – step 2.
The yield of 1 did not improve either with higher temperature (180 °C) or with a reaction time longer than 10 h. Both compounds 1 and 4 were isolated by column chromatography on silica gel in toluene/heptane (1:1 by vol.) and characterized (1H and 13C NMR, FT IR, elemental analysis, melting point, TLC). The R F values determined by TLC in the same solvent were 0.90 and 0.56 for 1 and 4, respectively. The tris(2-ethylhexyl) phosphite was used in 5 mol excess compared to both reaction components. It was more than the theoretical value of 3.5 mol (2 mols for ring closure + 1.5 mol for nitro compound) and sufficient amounts were present for reaction. Larger amounts of tris(2-ethylhexyl) phosphite caused problems in chromatographic separation of the reaction mixture and did not increase the yield of 1. The reported yields were determined on isolated amounts from column chromatography and, consequently, the true reaction yields may be slightly higher because of chromatographic loss. According to the FT IR (Figure ), 1H NMR (Figure ), and 13C NMR (Figure ) spectroscopy, the purity of the prepared compound 1 was excellent. We can conclude that the published Citation[6,8,9] syntheses of 1 can be now reduced to two steps: (a) nitration of 4,4′-diiodobiphenyl giving the compounds 3 and 5 and (b) carbazole ring closure by reaction of 3 and 5 with tris(2-ethylhexyl) phosphite to N-(2-ethylhexyl)-2,7-diiodocarbazole (1). The slightly lower overall yield of the new synthesis (13%) is compensated by the simplicity of the reaction process.
Next, we performed the carbazole ring closure (step 2) with compound 3 only to verify that the absence of 5 in the reaction mixture leads to a common Cadogan ring closure, i.e. without prevailing N-alkylation. The reaction of 3 with an excess of tris(2-ethylhexyl) phosphite in argon under the same reaction condition as above gave 4 as a major product (58%) and 1 as a minor product (3%). Again the 1H NMR (Figure ) and 13C NMR (Figure ) spectroscopy confirmed the high purity of the prepared compound 4.
Generally, the Cadogan ring closure reaction is usually performed with triethyl phosphite Citation[16,17]. We found that the triethyl phosphite mediated Cadogan ring closure on 3 led to the expected 4, whereas the same experiment on the mixture of compounds 3 and 5 afforded N-ethyl-2,7-diiodocarbazole (6) as a major product. The abridged preparation of 6 was of course interesting but utilization of this monomer in the syntheses of conjugated polymers would be limited. Short alkyl side chains are inconvenient from the solubility viewpoint for the final conjugated material. Monomers with longer or branched alkyl chains are required for syntheses of conjugated polymers in order to obtain soluble and high-molar-weight polymers Citation[18,19].
The moderate N-alkylation accompanying the Cadogan ring closure is known and it is probably caused Citation[20] by trialkyl phosphite and trialkyl phosphate that is present in the reaction mixture. It cannot explain our situation where nitro compound 5 apparently plays an important role. The carbazole N-ethylation was reported Citation[21] when o-nitrotoluene was added to triethyl phosphite mediated carbazole ring closure. According to the authors, the alkylation species such as triethyl N-(o-tolyl)phosphorimidate and diethyl N-(o-tolyl)phosphoramidate are because of the high temperature (170–210 °C) responsible for the N-alkylation process. The alkylation species were formed from triethyl phosphite and o-nitrotoluene during the reaction process. We can assume the same process in our procedure, where the alkylation species of tris(2-ethylhexyl) N-arylphosphorimidate and bis(2-ethylhexyl) N-arylphosphoramidate type can be formed from the 5 and tris(2-ethylhexyl) phosphite. As we mentioned above, the separation of products 1 and 4 was performed by column chromatography on silicagel in toluene/heptane (1:1 by vol.). This was followed by the application of gradient elution chromatography increasing the solvent polarity to toluene and acetone, which released more polar reaction intermediates from the column. These fractions were analyzed by GC-MS and reaction intermediates were determined as: 2-ethyl-1-hexanol, tris(2-ethylhexyl) phosphite, and tris(2-ethylhexyl) phosphate. We did not find species of phosphorimidate or phosphoramidate mentioned above. Perhaps they are not stable enough on silicagel (in the case of phosphorimidate it is probable) or during the GC-MS analysis. We continue to study the process.
4 Conclusion
In conclusion, we succeeded in a new two-step synthesis of N-(2-ethylhexyl)-2,7-diiodocarbazole, which is a suitable monomer for preparation of conjugated polymers for optoelectronics. It can be utilized directly in some polymerization procedures (Yamamoto, Sonogashira, Heck couplings, etc.) and/or as a convenient precursor for syntheses of the corresponding diboronic acid esters or trialkyl stannyl derivatives in other polymerization reactions (Suzuki or Stille couplings). We have shown the crucial role played by the aromatic nitro compound, 4-iodo-4′-nitrobiphenyl with 4,4′-diiodo-2-nitrobiphenyl in the reaction mixture. Using tris(2-ethylhexyl) phosphite simultaneous carbazole ring closure and N-alkylation was accomplished unlike the Cadogan ring closure, that is, without the nitro compound, where nonalkylated carbazole is formed as a main product.
Acknowledgments
The authors thank the Ministry of Education, Youth and Sports of the Czech Republic (grant No. 1M06031), and the Grant Agency of the Czech Republic (grant No. P106/12/0827) for financial support.
References
- Morin , J-F , Leclerc , M , Adès , D and Siove , A . 2005 . Polycarbazoles: 25 years of progress . Macromolecular Rapid Communications , 26 : 761 – 78 .
- Skotheim , TA and Reynolds , JR . 2007 . Handbook of conducting polymers , Boca Raton , FL : Taylor & Francis Group .
- Boudreault , P-LT , Beaupré , S and Leclerc , M . 2010 . Polycarbazoles for plastic electronics . Polymer Chemistry , 1 : 127 – 36 .
- Cimrová , V , Kmínek , I and Výprachtický , D . 2010 . Novel soluble fluorene-thienothiadiazole and fluorene-carbazole copolymers for optoelectronics . Macromolecular Symposia , 295 : 65 – 70 .
- Cadogan , JIG , Cameron-Wood , M , Mackie , RK and Searle , RJG . 1965 . The reactivity of organophosphorus compounds. Part XIX. Reduction of nitro-compounds by triethyl phosphite a convenient new route to carbazoles, indoles, indazoles, triazoles, and related compounds . Journal of the Chemical Society , : 4831 – 7 .
- Morin , J-F and Leclerc , M . 2001 . Syntheses of conjugated polymers derived from N-alkyl-2,7-carbazoles . Macromolecules , 34 : 4680 – 2 .
- Dierschke , F , Grimsdale , AC and Müllen , K . 2003 . Efficient synthesis of 2,7-dibromocarbazoles as components for electroactive materials . Synthesis , 16 : 2470 – 2 .
- Pan , X , Liu , S , Chan , HSO and Ng , S-C . 2005 . Novel fluorescent carbazoyl-pyridinyl alternating copolymers: synthesis, characterization, and properties . Macromolecules , 38 : 7629 – 35 .
- Výprachtický , D , Kmínek , I , Pavlačková , P and Cimrová , V . 2011 . A novel three-step synthesis of N-(2-ethylhexyl)-2,7-diiodocarbazole . Synthesis , 9 : 1472 – 6 .
- Freeman , AW , Urvoy , M and Criswell , ME . 2005 . Triphenylphosphine-mediated reductive cyclization of 2-nitrobiphenyls: a practical and convenient synthesis of carbazoles . Journal of Organic Chemistry , 70 : 5014 – 9 .
- Miyaura , N and Suzuki , A . 1995 . Palladium-catalyzed cross-coupling reactions of organoboron compounds . Chemical Reviews , 95 : 2457 – 83 .
- Výprachtický , D , Cimrová , V , Kmínek , I and Pavlačková , P . 2010 . Syntheses of conjugated polymers for photonics . Macromolecular Symposia , 295 : 94 – 9 .
- Hodson , HH . CCCXVII.—Studies in the diphenyl series. Part II. The nitration of diphthalylbenzidine . Journal of the Chemical Society , 1926 2384 – 6 .
- Harley-Mason , J and Mann , FG . 1940 . Quarterphenyl. Part I. Some dihydroxy-derivatives . Journal of the Chemical Society , : 1379 – 85 .
- Ponte , D . 1935 . Su alcuni nuovi derivati della 2,2’-dinitro-benzidina. Giornale di farmacia, di chimica e di scienze affini. 1934;83:347–50 . Chemical Abstracts , 29 : 29466
- Cadogan , JIG and Cooper , A . 1969 . Reduction of nitro- and nitroso-compounds by tervalent phosphorus reagents. Part III. Kinetic study of the triethyl phosphite reduction of 2-nitrosobiphenyl to carbazole . Journal of the Chemical Society (B) , : 883 – 5 .
- Cadogan JIG, Sears DJ, Smith DM, Todd MJ. Reduction of nitro- and nitroso-compounds by tervalent phosphorus reagents. Part V. Reduction of alkyl- and methoxy-nitrobenzenes, and nitrobenzene by trialkyl phosphites. Journal of the Chemical Society (C). 1969: 2813–9.
- Kmínek , I , Výprachtický , D , Kříž , J , Dybal , J and Cimrová , V . 2010 . Low-band gap copolymers containing thienothiadiazole units: synthesis, optical, and electrochemical properties . Journal of Polymer Science Part A: Polymer Chemistry , 48 : 2743 – 56 .
- Cimrová , V , Kmínek , I , Pavlačková , P and Výprachtický , D . 2011 . Low-bandgap donor-acceptor copolymers with 4,6-bis(3’-(2-ethylhexyl)thien-2’-yl)thieno[3,4-c][1,2,5]thiadiazole: synthesis, optical, electrochemical, and photovoltaic properties . Journal of Polymer Science Part A: Polymer Chemistry , 49 : 3426 – 36 .
- Puskas , I and Fields , EK . 1968 . Reactions of nitropolymethylbiphenyls . Journal of Organic Chemistry , 33 : 4237 – 42 .
- Tsunashima , Y and Kuroki , M . 1981 . Chemistry of carbazole. VI. On the formation of N-ethylcarbazoles in the cadogan reaction . Journal of Heterocyclic Chemistry , 18 : 315 – 8 .