Abstract
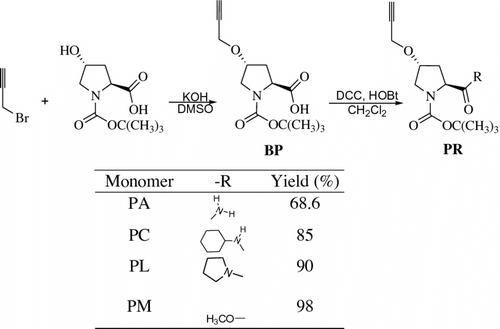
The novel acetylene monomers, L-proline-derived chiral propargylether (PA, PC, PL, and PM) were synthesized. The copolymerization of L-proline-based propargylether having amide group (PR) with N-(tert-butoxycarbonyl)-L-alanine-N-propargylamide (LA) by different unit ratio and the control of conformation of poly(LA-co-PR) were discussed. The copolymerization catalyzed by (nbd)Rh+[η 6-C6H5B−(C6H5)3] afforded copolymers with medium M n. The specific rotation of poly(LA88-co-PA12), poly(LA88-co-PC12), and poly(LA88-co-PL12) was 250°, 967°, and 1117°, respectively. The specific rotation of poly(LA-co-PC) ranged from −967° to −167°. Conformation change of poly(LA88-co-PR12) and poly(LA-co-PC) in different solvents was studied and it was found that the intramolecular hydrogen bonding in the side chain significantly contribute to the stabilization of helical conformation of the copolymers and that solvent polarity strongly affected the stability of helical conformation. The helical structure of poly(LA88-co-PL12) was affected by temperature change.
1. Introduction
The function of bioactive natural products such as nucleic acid, proteins and peptides, and polypeptides depends largely upon its conformation. So the study of conformation helps the understanding of the life forms Citation[1–4]. To date, the control of the conformation strongly attracts the attention of synthetic polymer scientists Citation[5–8]. Synthetic polymers can be tailor-made from a wide variety of compositions, molecular weights, dispersities, functionalities, shapes, etc. and their properties can be finely tuned for numerous applications in many fields such as chemistry, material, biology, medicine, and nanotechnology Citation[9–12].
The main chain of polyacetylene can form helical conformation in the solution when the interior and outer stimulus destroys its symmetry Citation[9,13]. Helically chiral polyacetylenes involve particular structures and morphology, which can be used in the several different fields on optical and stimuli-responsive materials, chiral cognition, biosensor Citation[14–17]. Poly(phenylacetylene) derivatives bearing a chiral pendant have been designed to form the consecutive one-handed twist of the main chain induced by chiral molecules Citation[18–20]. And poly(phenylacetylene) derivatives carrying amino acid moieties exhibit unique properties including self-assembling, formation of superhelical fibers, chirality transcription, and tuning the helicity by pH and solvent change Citation[13,21]. Helical chiral poly(N-propargylamide) has been studied to understand the relation between conformation and function of polymers Citation[22–25].
L-proline, a simple, abundant, and powerful chiral organocatalyst catalyzes the direct aldol reaction according to an enamine mechanism Citation[26]. The novel, optically active, cis-transoidal poly(phenylacetylene) bearing an L-proline residue pendant was prepared by polymerization and its chiroptical property was investigated using circular dichroism spectroscopy Citation[27]. The homopolymers of L-proline derivatives based propargylethers existed in no regulated higher order structure in solvents. Consequently, the copolymerization of L-proline-derived chiral propargylether with the L-alanine-derived N-propargylamide was accomplished and the chiroptical properties of the copolymers formed were examined Citation[28]. In the present study, we synthesized a novel acetylene monomer, L-proline-based propargylethers having amide group (PA, PC, PL, and PM in Scheme ). For tuning the conformation, poly(LA88-co-PR12) was copolymerized by N-(tert-butoxycarbonyl)-L-alanine-N-propargylamide (LA) to copolymerize with chiral propargylether (PR). The copolymers poly(LA-co-PC) of LA and PC with different unit ratios were formed and their conformation was also controlled in different solvents.
Scheme 1 Synthesis of L-proline based propargylether monomers PR.
2 Experimental
2.1 Measurements
1H and 13C NMR spectra were recorded in chloroform-d (CDCl3) on a Bruker DRX-400 spectrometer. IR spectra were measured using a Shimadzu FTIR-8100 spectrophotometer. The number- and weight-average molecular weights (M n and M w) of polymers were determined by gel permeation chromatography (GPC) on a Jasco Gulliver system (PU-980, CO-965, RI-930, and UV-1570) equipped with polystyrene gel columns (Shodex columns K804, K805, and J806), using THF as an eluent at a flow rate of 1.0 mL/min, calibrated by polystyrene standards at 40 °C. CD and UV–vis spectra were recorded in a quartz cell (thickness: 1 cm) at room temperature using a Jasco J810 spectropolarimeter. Melting points (mp) were measured on a Yanaco micro-melting point apparatus. Specific rotations ([α]D) were measured on a Jasco DIP-1000 digital polarimeter with a sodium lamp as a light source (wavelength 598 nm, temperature 20 °C).
2.2 Materials
The solvents were distilled by usual methods prior to use. Propargylamine (Acros Oganics), propargyl bromide (Sigma–Aldrich Co.), trans-N-(tert-Butoxycarbonyl)-4-hydroxy-L-proline (Boc-Hyp-OH; Sigma–Aldrich Co.), 1-hydroxybenzotriazole monohydrate (HOBt; Acros Oganics), 1-(3,3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (DCC; Acros Oganics) and (bicyclo[2,2,1]hepta-2,5-diene)chlororhodium(I) dimmer {[Rh(nbd)Cl]2, nbd = 2,5-norbornadiene; Sigma–Aldrich Co.}, were used as purchased without purification. (nbd)Rh+[η 6-C6H5B−(C6H5)3] was prepared by the reaction of [(nbd)RhCl]2 with NaB(C6H5)4 as described in the literature Citation[29]. LA was synthesized according to the reported method Citation[30].
2.3 Monomer synthesis
Preparation of Boc-propargyloxyproline (BP) is described as a typical procedure. In a 250 mL Morton three-neck round-bottomed flask equipped with a mechanical stirrer, finely powdered 82% KOH (41 g, 0.6 mol) was dissolved in DMSO (80 mL) under an atmosphere of nitrogen. After the mixture was stirred at ambient temperature for 15 min, the apparatus was cooled to 0 °C with an ice-water bath. After the mixture was stirred for 10 min, Boc-Hyp-OH (18 g, 79 mmol) was added in one portion. After being stirred for 60 min, the completely homogeneous solution was treated with one portion of propargyl bromide (44.6 g, 0.3 mol). After being stirred at 0 °C for 15 min, the reaction mixture was warmed to ambient temperature for 4 h. The reaction was monitored by the following procedure. The reaction mixture (0.5 mL) was aliquoted to a 4 mL glass vial and diluted with water (1 mL). The aliquot was acidified with 1 M aqueous KHSO4 (0.5 mL) to pH 2–3 (pH paper). Diethyl ether (2 mL) was added to the acidic mixture. The organic phase was analyzed by thin layer chromatography (TLC) (9:1 CH2Cl2/MeOH) using I2 detection. After the reaction was judged complete by TLC after 4 h, the reaction mixture was poured into water (300 mL). The Morton flask was rinsed with an additional portion of water (50 mL), and the aqueous wash was transferred into the reaction mixture. After being stirred at ambient temperature for 5 min, the suspension was washed with diethyl ether (2 × 200 mL). The aqueous phase was acidified with 87% concentrated H3PO4 to pH 2–3 (pH paper). This solution was then extracted with diethyl ether (2 × 200 mL). The combined ether layers were washed with water (2 × 100 mL) and saturated aqueous NaCl (2 × 50 mL). The organic layer was dried over anhydrous MgSO4 for 30 min, filtered, and concentrated in vacuo overnight in 64.7% yield, white solid, mp: 79 °C, [α]D = 36.7° (c = 0.3 g/dL in CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.29 and 1.37 [C(CH3)3, m, 9H], 1.93–1.99 and 2.29–2.35 (CH2, 2H), 2.46 (C≡CH, 1H), 3.39–3.43 (CCH2N, 2H), 4.02–4.07 (OCH, 1H),4.14 (C≡CCH2, 2H), 4.19 (C=OCH, 1H), 12.62 (COOH, 1H). 13C NMR (100 MHz, CDCl3) δ 27.8, 28.0, 34.7, 35.4, 51.2, 51.6, 55.7, 57.2, 57.5, 75.4, 76.2, 77.2, 79.0, 80.3, 153.0, 153.6, 173.5, 174.0. IR (cm−1, KBr): 3473, 3258, 2980, 2870, 2716, 2580, 2129 (C≡C), 1750 (C=O), 1640 (C=O), 1558, 1479, 1433, 1369, 1263, 1082, 992, 942, 910, 853, 695, 663, 554.
Preparation of PA is described as a typical procedure. In CH2Cl2 (50 mL), BP (4.5 g, 19.3 mmol) was dissolved, and 1-hydroxybenzotriazole monohydrate (HOBt) (3.6 g, 23.5 mmol) and 1-(3, 3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (DCC) (4.5 g, 23.5 mmol) were then added with ice-cooling. The temperature was gradually increased, and the mixture was then stirred at room temperature overnight. The reaction solution was again ice-cooled, 25% aqueous ammonia (5 mL) was added, and stirring was continued with ice-cooling for 30 min and then at room temperature for 30 min. Acetonitrile (50 mL) was added to the reaction mixture, and an insoluble substance was removed by filtration. The filtrate was concentrated in vacuo and purified by silica gel column chromatography (developing solvent; hexane–EtOAc = 4:1–1:5).
PA: yield 68.6%, white solid, mp: 89 °C, [α]D = −63.3° (c = 0.3 g/dL in CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.44 [C(CH3)3, m, 9H], 1.60–1.69 and 2.29–2.35 (CH2, 2H), 2.42–2.43 (C≡CH, 1H), 3.47–3.51 (CCH2N, 2H), 4.02–4.07 (OCH, 1H),4.13 (C≡CCH2, 2H), 4.19 (C=OCH, 1H), 5.38, 5.89 and 6.78 (C=ONH 2 , 2H). 13C NMR (100 MHz, CDCl3) δ 28.3, 33.6, 33.9, 49.2, 51.6, 56.5, 58.1, 59.5, 74.7, 75.8, 79.3, 80.9, 154.5, 155.7, 173.9, 175.1. IR (cm−1, KBr): 3377, 3224, 3203, 3001, 2977, 2941, 2891, 2851, 2110 (C≡C), 1690, 1667, 1478, 1425, 1366, 1176, 1142, 1088, 981, 928, 856, 774, 743, 713, 647, 547.
PC: Yield 85%, white solid, mp: 91 °C, [α]D = −61.7° (c = 0.3 g/dL in CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.15, 1.12–1.39, 1.59, 1.66 (C6H10, 10H), 1.43 [C(CH3)3, m, 9H], 1.84 (CH2, 2H), 2.41–2.42 (C≡CH, 1H), 3.47–3.48 (C=OCH, 1H), 3.72 (NCH, 1H), 4.09–4.13 (C≡CCH2, 2H), 4.31 (CCH 2 N, 2H), 5.78 and 6.85 (C=ONH, 2H). 13C NMR (100 MHz, CDCl3) δ 24.5, 24.8, 25.4, 28.3, 32.7, 33.1, 33.9, 47.9, 51.5, 56.5, 58.5, 60.0, 74.7, 75.7, 79.3, 80.6, 154.7, 155.7, 170.2, 171.1. IR (cm−1, KBr): 3311, 3064, 2663, 2972, 2934, 2878, 2854, 2113 (C≡C), 1697, 1657, 1545, 1415, 1387, 1262, 1229, 1170, 1139, 1091, 1004, 908, 766.
PL: yield 90%, white solid, mp: 101 °C, [α]D = −28.3° (c = 0.3 g/dL in CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.37 and 1.42 [C(CH3)3, m, 9H], 1.80–1.85 and 1.93–1.96 (CH 2 , 4H), 2.08–2.11 and 2.29–2.35 (CH 2 , 2H), 2.42–2.43 (C≡CH, 1H), 3.36–3.40 and 3.45–3.48 (CH 2 NCH 2 , 4H), 3.51–3.55 and 3.64–3.65 (CCH2N, 2H), 4.10–4.16 (C≡CCH2, 2H), 4.33–4.56 (C=OCH and OCH, 2H). 13C NMR (100 MHz, CDCl3) δ 24.0, 26.1, 28.3, 28.4, 35.3, 36.3, 45.9, 46.0, 46.2, 51.2, 51.8, 56.3, 56.4, 56.5, 74.5, 74.6, 79.4, 79.6, 153.6, 154.3, 170.6, 170.8. IR (cm−1, KBr): 3204, 3008, 2973, 2953, 2872, 2120 (C≡C), 1691, 1647, 1447, 1413, 1362, 1340, 1162, 1132, 1092, 1076, 1006, 911, 890, 861, 770, 717, 550.
PM: yield 98%, yellow oil, [α]D = −26.7° (c = 0.3 g/dL in CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.38 and 1.43 [C(CH3)3, m, 9H], 2.03–2.10 and 2.35–2.36 (CH2, 2H), 2.42–2.43 (C≡CH, 1H), 3.60–3.63 (CCH2N, 2H), 3.71–3.72 (–OCH3, 3H), 4.12–4.15 (C≡CCH2, 2H), 4.29–4.33 (C=OCH and OCH, 2H). 13C NMR (100 MHz, CDCl3) δ 28.2, 28.3, 35.3, 36.5, 51.0, 51.6, 52.0, 52.2, 56.3, 57.4, 57.8, 74.8, 75.7, 79.2, 80.2, 153.6, 154.2, 173.2, 173.5. IR (cm−1, KBr): 3265, 2980, 2954, 2118 (C≡C), 1751, 1702, 1541, 1404, 1369, 1259, 1207, 1163, 1129, 1092, 980, 918, 898, 775, 679.
2.4 Copolymerization procedure
As illustrated in Scheme , all the polymerizations were carried out in a Schlenk tube equipped with a three-way stopcock under nitrogen. A solution of LA (0.88 mmol) and PC (0.12 mmol) in CHCl3 (5 mL) was added to (nbd)Rh+[η 6-C6H5B−(C6H5)3] (10.6 mg, 0.02 mmol), and the resulting mixture was vigorously stirred. It was kept in a water bath at 30 °C for 12 h. The resulting mixture was poured into n-hexane (200 mL) to precipitate a copolymer. It was separated by filtration, and dried under reduced pressure. Yield: 149 mg (66%).
Scheme 2 Copolymerization of LA and PC with different unit ratios.
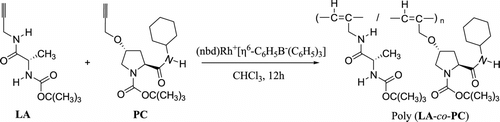
Poly(LA88-co-PA12): 1H NMR (400 MHz, CDCl3): δ 1.29–1.43 (broad s), 3.41–3.51 (broad s), 4.03 (s), 4.14 (s), 4.30 (broad s), 4.86 (s), 6.35 (s, 1H, –CH=C); IR (cm−1, KBr): 3320, 2980, 2935, 1660 (C=O), 1534, 1358, 1311,1245, 1178.
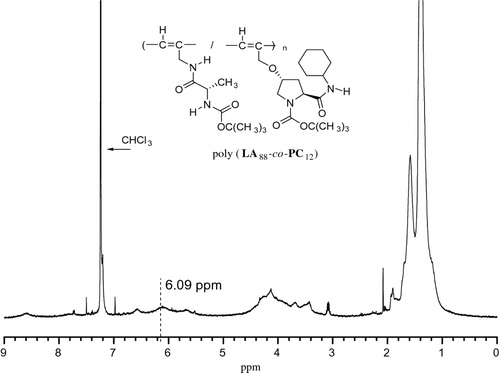
Poly(LA88-co-PC12): 1H NMR (400 MHz, CDCl3) in Figure : δ 1.39 (broad s), 3.41–3.45 (broad s), 3.69 (broad s), 3.85 (s), 3.97 (s), 4.09–4.03 (broad s), 4.13 (broad s), 4.34–4.42 (s), 6.10 (broad s, 1H, –CH=C); IR (cm−1, KBr): 3340, 3299, 2979, 2935, 1675 (C=O), 1536, 1452, 1388, 1367, 1301, 1251,1170, 1072.
Figure 1 1H NMR spectra of chloroform solution of poly(LA88-co-PC12).
Poly(LA88-co-PL12): 1H NMR (400 MHz, CDCl3): δ 1.39–1.43 (broad s), 3.44 (broad s), 3.83–4.34 (broad t), 3.71 (broad s), 6.10 (broad s, 1H, –CH=C), 3.60 (broad s); IR (cm−1, KBr): 3324, 2979, 2935, 2362, 1746, 1658 (C=O), 1521, 1452, 1367, 1247, 1166, 1052.
3 Results and discussion
3.1 Monomer synthesis
A practical synthesis of BP was accomplished by alkylation of Boc-hydroxyproline with propargyl bromide (Scheme ), employing KOH and DMSO as base and solvent. This process was executed under anhydrous conditions and was driven to completion using 400 mol% alkylating reagent (propargyl bromide). Use of H3PO4 in the workup avoided the precipitation of solids that complicated product isolation when KHSO4 was employed as the reagent for acidification. Purification of the crude BP was accomplished via the cyclohexylamine salt in order to remove an unknown byproduct. No chromatography was required in the syntheses of the alkylation reaction.
Acylation of achiral amine and esterification of MeOH with BP were successfully accomplished using DCC/HOBt in CH2Cl2 (Scheme ). Purification of the crude PR was accomplished by silica gel column chromatography (developing solvent; hexane–EtOAc = 4:1–1:5).
3.2 Copolymerization
Noyori et al. Citation[31] reported that a rhodium catalyst could efficiently catalyze the polymerization of monosubstituted acetylenes through an insertion mechanism to give mainly cis-transoidal polyacetylenes. Thus, a rhodium catalyst, (nbd)Rh+[η 6-C6H5B−(C6H5)3], was investigated as a catalyst in our study. The monomers were completely consumed within 12 h to afford polymers with medium molecular weight.
The copolymerization of L-proline derivatives based PR and N-(tert-butoxycarbonyl)-L-alanine-N-propargylamide (LA) with the rhodium catalyst, (nbd)Rh+[η 6-C6H5B−(C6H5)3], gives yellow polyacetylenes. 1H NMR spectrum (no data) analyzed there is a broad signal at δ ∼6.1 ppm, which is absent in the spectrum of its monomer. This signal is assigned to the olefinic proton of the main chain having the cis-transoidal structure (this portion will be reported as the other article).
Scheme and Table summarize the conditions and results of the copolymerization of LA with PR catalyzed by (nbd)Rh+[η 6-C6H5B−(C6H5)3] in CHCl3 at 30 °C for 12 h. The color of the n-hexane-insoluble part of 12% PA and 88% LA was white with low M n (2337) obtained. 1H NMR spectrum analyzed that the protons of two acetylene monomers existed, it was shown that the copolymerization of the two acetylene monomers (PA and LA) was not complete. On the other hand, monomers LA with PC and PL satisfactorily underwent copolymerization to afford the corresponding copolymers with the M n (11,492, 6993), which were soluble in THF, CH2Cl2, and CHCl3, but insoluble in n-hexane. 1H NMR spectrum analyzed that the protons of two acetylene monomers did not exist. At the same time, the copolymerization of 12% PM with 88% LA was also investigated. But no n-hexane-insoluble polymer was obtained. The color of the homopolymers of LA, poly(LA88-co-PC12), and poly(LA88-co-PL12) was yellow, and that of poly(LA88-co-PA12) became white.
Table 1. Homopolymerization of LA and copolymerization of LA with PR.a
3.3 Copolymerization of two different acetylene monomers LA and PC
Scheme and Table summarize the conditions of results of the copolymerization of LA with PC catalyzed by (nbd)Rh+[η 6-C6H5B−(C6H5)3] in CHCl3 at 30 °C for 12 h. The different acetylene monomers LA and PC underwent copolymerization to afford the corresponding copolymers with the M n in the range from 11,492 to 2954, which were soluble in THF, CH2Cl2, and CHCl3, but insoluble in n-hexane. With the improvement of PC in the unit ratio, M n and specific rotation of poly(LA-co-PC) decreased and it was obvious to the specific rotation: −967 (88:12), −683 (75:25), and −383 (68:32). The color of poly(LA) was yellow and that of poly(PC) was orange and that of the copolymers was between yellow and orange. The copolymerization of LA with 12% of PC is more satisfying because it obtained the highest M n (11,492) and specific rotation (−967) than the homopolymerization of LA.
Table 2. Copolymerization of LA with PC.a
3.4 Conformation change of poly(LA88-co-PR12) in different solvents
By analyzing Scheme , it is seen that monomer PA exists with two N–H bonds, and the CD spectrum of poly(LA88-co-PA12) existed in the transition state between helical conformation at 400 nm and random structure at 320 nm in CHCl3. When CH3OH was added into CHCl3, the CD signal at 400 nm disappears and only one CD signal exists at 320 nm in Figure . This means that the helical conformation disappeared and the intramolecular hydrogen bond between the copolymers was destroyed by –OH of CH3OH.
Figure 2 (A) CD and (B) UV–vis spectra of poly(LA88-co-PA12) measure in (a) CHCl3 and (b) CHCl3/MeOH = 1:1 at room temperature, c = 3 × 10−4 mol/L.
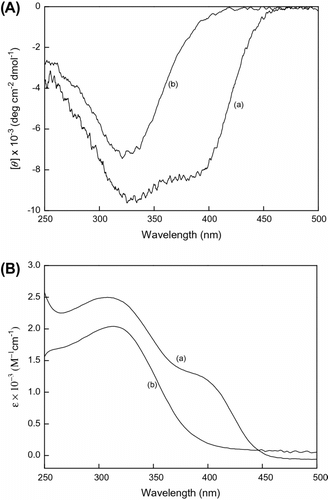
Figure shows the CD and UV–vis spectral changes of poly(LA88-co-PC12) in different solvents. The spectra were measured in CHCl3, CHCl3/THF mixed solvents with the volume composition varying from 1/0.25 to 1/10. Both the CD signal and UV–vis absorption at 400 nm were slightly increased by the addition of THF to CHCl3, and the most large absorption was little red-shifted compared with in CHCl3. But the CD signal and UV–vis absorption at 400 nm disappeared in MeOH, which indicates the destruction of the helical structure. And CH3OH in the solvent could destroy the helical conformation of poly(LA88-co-PC12) as shown in Figure .
Figure 3 (A) CD and (B) UV–vis spectra of poly(LA88-co-PC12) measure in (a) CHCl3, (b) CHCl3/THF = 1/0.25, (c) CHCl3/THF = 1/1, (d) CHCl3/ THF = 1/10, and (e) MeOH at room temperature, c = 3 × 10−4 mol/L.
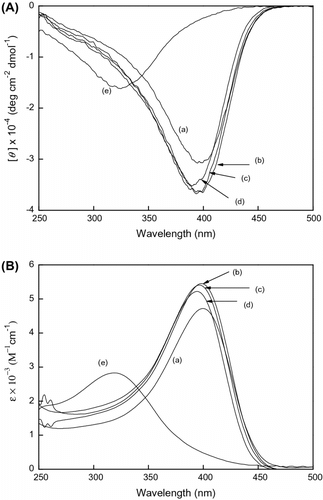
Figure shows the conformation change of poly(LA88-co-PL12) in a different solvent. The conformation existed between the helical and random structure in THF. When CH3OH was added into CHCl3, the helical conformation was destroyed. From the analyses of the three copolymers, CH3OH would destroy the helical conformation as shown in Figures and . The conformation in THF was unstable and was located in the transition state between the helical conformation at 400 nm and the random coil at 320 nm.
Figure 4 (A) CD and (B) UV–vis spectra of poly(LA88-co-PL12) measure in (a) CHCl3, (b) THF, (c) CHCl3/MeOH = 1/1, and (d) CHCl3/MeOH = 1/2 at room temperature, c = 3 × 10−4 mol/L.
3.5 Conformation change of poly(LA-co-PC) in different solvents
We had synthesized the novel acetylene monomer PC and investigated the secondary structure of poly(PC). But it had no regulated higher order structure. Thus for reconstruction of helical conformation, we study whether or not to successfully complete the copolymerization of the chiral propargylether monomer PC with N-(tert-butoxycarbonyl)-L-alanine N-propargylamide LA by different unit ratio and analyze the chiroptical properties of the copolymers in different solvents. With improvement of the content of PC in ploy(LA-co-PC), the lack of the copolymer on hydrogen bond became obvious and led to the disappearance of helical conformation.
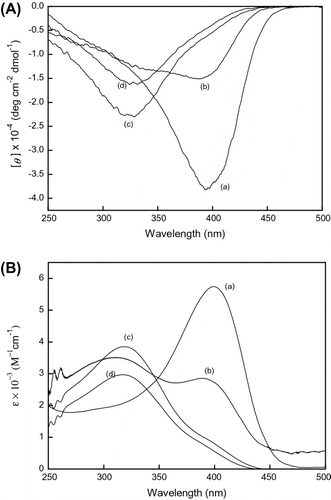
The conformation of poly(LA75-co-PC25) was unstable and was located in the transition state between the conformation at 396 nm and that at 320 nm. In Figure , the CD signal at 400 nm of poly(LA88-co-PC12) is found to disappear by the addition of MeOH. So we studied the conformation change of poly(LA75-co-PC25) in mixed solvents CHCl3/MeOH (1:1 and 1:0.3) and the results are shown in Figure . When MeOH was added to CHCl3 by the volume ratio (1:1), the CD signal at 396 nm disappeared and the CD signal at 320 nm got larger than that in CHCl3. The UV–vis largest absorption existed in 318 nm. For investigating the effect of the volume of MeOH on the conformation, we decreased the volume of MeOH in CHCl3/MeOH by the volume ratio (1:0.3) and found that the addition of little volume of MeOH could change the conformation. But the CD signal and UV–vis absorption almost had no change in mixed solvents. We can conclude that the polar solvent MeOH destroys the helical conformation in CHCl3.
Figure 5 (A) CD and (B) UV–vis spectra of poly(LA75-co-PC25) measure in (a) CHCl3, (b) CHCl3/MeOH = 1:1, and (c) CHCl3/MeOH = 1:0.3 at room temperature, c = 3 × 10−4 mol/L.
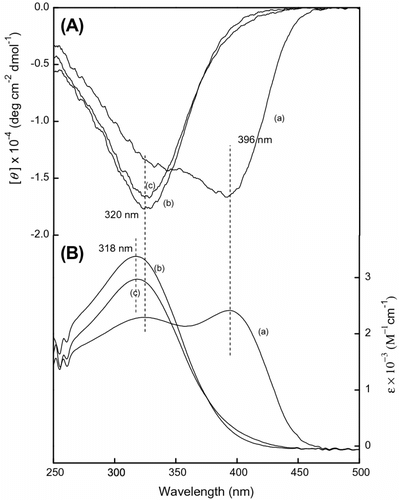
Figure shows the CD and UV–vis spectral changes of poly(LA68-co-PC32) with the solvent. The spectra were measured in CHCl3 and CHCl3/MeOH (1:1 and 1:11). Compared with the CD and UV–vis spectra of poly(LA75-co-PC25), both the CD signal and UV–vis absorption at 400 nm did not exist in the conformation of poly(LA68-co-PC32) in solvents and the new CD signal at 329 nm and UV–vis absorption at 318 nm appeared in the spectra. When the polar solvent MeOH was added to CHCl3 by the volume ratio (1:1), the CD signal at 329 nm almost had no change and the UV–vis absorption at 318 nm got larger. And when kept on an increasing mode by the addition of MeOH to CHCl3 (11:1), the CD signal and UV–vis absorption evidently decreased and the most large absorption was little red-shifted.
Figure 6 (A) CD and (B) UV–vis spectra of poly(LA68-co-PC32) measure in (a) CHCl3, (b) CHCl3/MeOH = 1:1, and (c) CHCl3/MeOH = 1:11 at room temperature, c = 3 × 10−4 mol/L.
3.6 Variable temperature CD of poly(LA88-co-PL12)
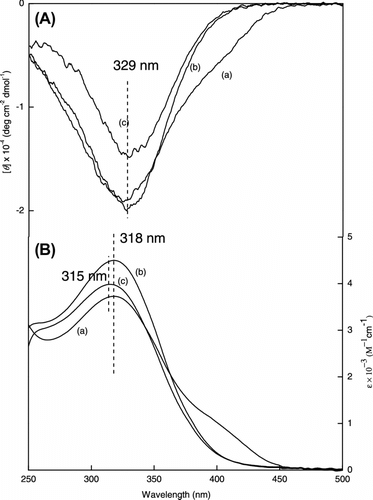
The helical conformation of poly(LA88-co-PL12) is located at 396 nm. To examine the thermal effect on the conformation transformation, we measured CD and UV–vis spectra of poly(LA88-co-PL12) in CHCl3 at various temperatures. The CD spectrum of the copolymer showed remarkably decreased molar ellipticity with increasing temperature at 396 nm except below 0 °C. However, the shape did not change at 320 nm (Figure ). More specifically, the intensity of CD signal at 0 °C was slightly higher than that at −10 °C and it means that lower temperature (below 0 °C) is not favorable to form helical conformation of poly(LA88-co-PL12). At 30 °C, the CD spectrum of the copolymer has two CD signals of the same intensity at 320 and 396 nm. And in the range 50–60 °C the CD signal at 396 nm disappears and only one CD signal exists at 320 nm. The conformation of poly(LA88-co-PL12) remarkably changed with changing temperature and PL obtains the leadership in the competition on the conformation of poly(LA88-co-PL12) in CHCl3 with the increase in temperature. From UV–vis spectra we can also clearly see that the change of the role between PL and LA. On the other hand, poly(LA88-co-PL12) was less conjugated in CHCl3 by the increase in temperature.
Figure 7 Variable temperature CD (A) and UV–vis (B) spectra [C] dependence of the second Cotton effect of poly (LA88-co-PL12) at ∼400 nm on the temperature measure in CHCl3 (c = 3 × 10−4 mol/L).
![Figure 7 Variable temperature CD (A) and UV–vis (B) spectra [C] dependence of the second Cotton effect of poly (LA88-co-PL12) at ∼400 nm on the temperature measure in CHCl3 (c = 3 × 10−4 mol/L).](/cms/asset/a9e1e41a-4c05-4bbd-952b-0d168f5378a9/tdmp_a_747150_o_f0007g.gif)
4 Conclusion
The novel acetylene monomers, L-proline-derived chiral propargylether (PA, PC, PL, and PM) were synthesized and copolymerized with the L-alanine-derived N-propargylamide (LA). The conformation control of the formed copolymers was studied by different compositions, solvents, and temperatures. It was confirmed that the copolymerization of 12% PM with LA did not obtain n-hexane insoluble copolymer. And the copolymerization of 12% PA with LA did not completely react as a white powder, which had two monomers examined by 1H NMR. The copolymerization of 12% PC and PL with LA (M n = 11,492, 6993) was successful. Poly(LA88-co-PC12) effectively takes helical conformations in CHCl3. The CD signal of poly(LA75-co-PC25) exists at 400 and 320 nm in CHCl3. Poly(LA68-co-PC32) takes the random coil at 320 nm in CHCl3. The CD signal and UV–vis absorption are red-shifted by the addition of MeOH to CHCl3. The helical conformation disappeared and the intramolecular hydrogen bond between copolymers was destroyed by CH3OH. The novel structural feature of the copolymers was that a conjugated polyacetylene backbone was wrapped up with a coat of Boc-L-proline as open–active site and the copolymers would form helical conformation in solvents. The copolymers would allow to be applied to chiral resolution and biosensors.
Acknowledgments
The work described in this paper was partially supported by National Natural Science Foundation of China (21104097) and Natural Science Foundation of Guangdong Province (9451027501003025).
References
- Hill , KK , Roemer , SC and Churchill , MEA . 2012 . Structural and functional analysis of domains of the progesterone receptor . Mol. Cell Eedocrinol. , 348 : 418 – 429 .
- Schulman , BA . 2011 . Twists and turns in ubiquitin-like protein conjugation cascades . Protein Sci. , 20 : 1941 – 1954 .
- Carlo , B , Marco , P and Angela , DS . 2011 . Structural characterization of recombinant therapeutic proteins by circular dichroism . Curr. Pharm. Biotechnol. , 12 : 1508 – 1516 .
- Yang , W . 2011 . Nucleases: diversity of structure, function and mechanism . Q. Rev. Biophys. , 44 : 1 – 93 .
- Ma , B and Nussinov , R . 2010 . Enzyme dynamics point to stepwise conformational selection in catalysis . Curr. Opin. Chem. Biol. , 14 : 652 – 659 .
- Andrew , TL and Swager , TM . 2011 . Structure-property relationships for exciton transfer in conjugated polymers . J. Polym. Sci. Pol. Phys. , 49 : 476 – 498 .
- Rabotyagova , OS , Cebe , P and Kaplan , DL . 2011 . Protein-based block copolymers . Biomacromolecules. , 12 : 269 – 289 .
- Hashidzume , A and Harada , A . 2011 . Recognition of polymer side chains by cyclodextrins . Polym. Chem. , 2 : 2146 – 2154 .
- Akagi , K . 2009 . Helical polyacetylene: asymmetric polymerization in a chiral liquid-crystal field . Chem. Rev. , 109 : 5354 – 5401 .
- Förster , S and Antonietti , M . 1998 . Amphiphilic block copolymers in structure-controlled nanomaterials hybrids . Adv. Mater. , 10 : 195 – 217 .
- Park , C , Yoon , J and Thomas , EL . 2003 . Enabling nanotechnology with self-assembled block copolymer patterns . Polymer , 44 : 6725 – 6760 .
- Förster , S and Konrad , M . 2003 . From self-organizing polymers to nano- and biomaterials . J. Mater. Chem. , 13 : 2671 – 2688 .
- Lam , JWY and Tang , BZ . 2005 . Functional polyacetylenes . Acc. Chem. Res. , 38 : 745 – 754 .
- Yashima , E . 2010 . Synthesis and structure determination of helical polymers . Polym. J. , 42 : 3 – 16 .
- Yashima , E , Maeda , K and Iida , H . 2009 . Helical polymers: synthesis, structures, and functions . Chem. Rev. , 109 : 6102 – 6211 .
- Liu , K , Yu , Z and Liu , J . 2009 . Molecular shapes of monosubstituted polyacetylenes in their liquid crystalline phases . Macromol. Chem. Phys. , 210 : 708 – 716 .
- Goh , M , Matsushita , S and Akagi , K . 2010 . From helical polyacetylene to helical graphite: synthesis in the chiral nematic liquid crystal field and morphology-retaining carbonisation . Chem. Soc. Rev. , 39 : 2466 – 2476 .
- Kumaki , J , Sakurai , S and Yashima , E . 2009 . Visualization of synthetic helical polymers by high-resolution atomic force microscopy . Chem. Soc. Rev. , 38 : 737 – 746 .
- Yashima , E and Maeda , K . 2008 . Chirality-responsive helical polymers . Macromolecules. , 41 : 3 – 12 .
- Maeda , K , Mochizuki , H , Watanabe , M and Yashima , E . 2006 . Switching of macromolecular helicity of optically active poly(phenylacetylene)s bearing cyclodextrin pendants induced by various external stimuli . J. Am. Chem. Soc. , 128 : 7639 – 7650 .
- Li , BS , Kang , SZ , Cheuk , KKL , Wan , LJ , Ling , LS , Bai , CL and Tang , BZ . 2004 . Self-assembling of helical poly(phenylacetylene) carrying L-valine pendants in solution, on mica substrate, and on water surface . Langmuir. , 20 : 7598 – 7603 .
- Shiotsuki , M , Sanda , F and Masuda , T . 2011 . Polymerization of substituted acetylenes and features of the formed polymers . Polym. Chem. , 2 : 1044 – 1058 .
- Qu , J , Shiotsuki , M , Sanda , F and Masuda , T . 2007 . Synthesis and properties of helical polyacetylenes carrying cholesteryl moieties . Macromol. Chem. Phys. , 208 : 823 – 832 .
- Sanda , F , Fujii , T , Tabei , J , Shiotsuki , M and Masuda , T . 2008 . Synthesis of hydroxy group-containing poly(N-propargylamides): examination of the secondary structure and chiral-recognition ability of the polymers . Macromol. Chem. Phys. , 209 : 112 – 118 .
- Zhao , H , Sanda , F and Masuda , T . 2004 . Transformation of helical sense of poly(N-propargylamides) controlled by competition between structurally different enantiomeric amino acids . Macromolecules. , 37 : 8888 – 8892 .
- List , B . 2002 . Proline-catalyzed asymmetric reactions . Tetrahedron. , 58 : 5573 – 5590 .
- Kawamura , H , Takeyama , Y , Yamamoto , M , Kurihara , H , Morino , K and Yashima , E . 2011 . Chirality responsive helical poly(phenylacetylene) bearing L-proline pendants . Chirality. , 23 ( Suppl 1 ) : E35 – E42 .
- Dai , J , Ha , CY , Chang , DL and Shen , MM . 2007 . Helical conformation of the copolymers of L-proline derivatives based propargylether and chiral N-propargylamide . Polymer. , 48 : 5696 – 5701 .
- Schrock , RR and Osborn , JA . 1970 . π-Bonded complexes of the tetraphenylborate ion with rhodium(I) and iridium(I) . Inorg. Chem. , 9 : 2339 – 2343 .
- Gao , G , Sanda , F and Masuda , T . 2003 . Synthesis and properties of amino acid-based polyacetylenes . Macromolecules. , 36 : 3932 – 3937 .
- Kishimoto , Y , Itou , M , Miyatake , T , Ikariya , T and Noyori , R . 1995 . Polymerization of monosubstituted acetylenes with a zwitter ionic rhodium(I) complex, Rh+(2,5-norbornadiene) [(eta(6)-C6H5B-(C6H5)(3)] . Macromolecules. , 28 : 6662 – 6666 .