
Abstract
Optically active polyacetylenes (LPA and DPA) and racemic polyacetylenes (RPA) were prepared by the polymerization of N-propioloyl-tyrosine methyl esters that derived from chiral and racemic tyrosine. All the polymers were characterized by Fourier transform infrared spectroscopy, 1H NMR, 13C NMR, gel permeation chromatography, thermogravimetric analysis, UV–vis spectroscopy, and circular dichroism spectroscopy; furthermore, the infrared emissivity values were also investigated. Both LPA and DPA possessed the helical conformations and a higher degree of intra- and interchain hydrogen bonds compared with RPA, which presented irregular polymer chain. The thermal stability of LPA and DPA was higher than that of RPA due to the helical conformations, which could be easy to form a high degree of intra- and interchain hydrogen bonds in the structure. Therefore, LPA and DPA exhibited lower infrared emissivity values (8–14 μm) than that of RPA, which resulted from the helical conformations and a higher degree of intra- and interchain hydrogen bonds.
Introduction
In recent years, materials with low infrared emissivity have attracted much attention due to their widely application prospects in thermal control technology and infrared stealthy material.[Citation1] Various novel materials such as strongly magnetic materials, metallic thin films, nanosized semiconductor particles, and inorganic–organic composites are very widely applied in the field of infrared emissivity.[Citation2–5]
Organic polymers possess unique properties such as low density, tractability, and corrosion resistance. Most importantly, their adjustable molecular structure provide the possibility of changing the infrared emissivity according to applied conditions.[Citation6] Thus, macromolecular materials can meet the demand of the growing applications in infrared emissivity control. Meanwhile, optically active polymers have attracted great attention owing to the common advantages of any other macromolecules and their characteristic features such as ordered helical structure, multiple inter chain interaction, and adjustable optical parameter.[Citation7–10] Consequently, their characteristic features provide the potential applications in electrical conductivity, asymmetric synthesis, molecular recognition, liquid crystalline materials for display, and infrared emissivity materials.[Citation11–15] Among the majority of optically active polymers, polyacetylene is very interesting since it is a π-conjugated polymer which possesses a symmetric chain.[Citation16] Polyacetylene and its derivatives possess unique properties such as helix inversion, semi-conductivity, electrical conductivity, and nonlinear optical properties.[Citation17,18] The symmetry of the polymer chain, however, can be damaged by connecting chiral pendants to the polyacetylene backbone. As a result, it can induce the polymer chain to spiral in a helical conformation.[Citation19–21] Moreover, a variety of polyacetylenes containing mesogenic and chromophoric groups with ester,[Citation22,23] ether,[Citation24–27] amine,[Citation28–30] thio,[Citation31,32] and cyano [Citation33,34] functionalities have been successfully synthesized and potentially used as liquid crystals, mesomorphic luminescent and conductive materials, and so on. Natural amino acids have been generally applied in the preparation of optically active materials as one of the most significant chiral resources.[Citation35–38] Furthermore, macromolecules containing chiral amino acids pendants could form the ordered secondary structure that have enhanced properties. However, as far as we know, there are no reports referred to the investigation of infrared emissivity properties of optically active polyacetylenes carrying amino acid pendants.
In recent research of polyacetylenes, the functional pendants in the polymers were mostly apart from the polyacetylene backbone by saturated aliphatic spacers or aromatic rings.[Citation22–34] However, we had an interest in directly connecting chiral amino acid pendants to the polyacetylene backbone, and in this study, we made our best efforts to polymerize monosubstituted propiolamides (HC≡CCONHR), in which the triple bond was directly attaching to the carbonyloxy group. We used propiolic acid and tyrosine methyl esters in the synthesis of the monomer containing tyrosine derivatives as a functional group. The rhodium catalyst [Rh(nbd)Cl]2 was used in the polymerization of the monomer. The polymers with helical structure and a higher degree of hydrogen bonds (LPA and DPA) possessed a lower infrared emissivity value compared with the racemic one (RPA). According to the effect of helical conformations and intra- and interchain hydrogen bonds on the infrared emissivity, a novel infrared low emissivity material was provided to investigate.
Experimental
Materials
l-tyrosine, dl-tyrosine, anhydrous magnesium sulfate, and anhydrous sodium bicarbonate were purchased from Shanghai Chemical Reagent Company and used as received without further purification. d-tyrosine, propiolic acid, 4-dimethylaminopyridine (DMAP), and dicyclohexylcarbodiimide (DCC) were the products of Aladdin. Tetrahydrofran (THF) and triethylamine (TEA) were purchased from Shanghai Chemical Reagent Company and purified by distillation prior to use. Thionyl chloride, methanol, hexane, ethyl acetate, N,N-dimethylformamide (DMF), N,N-dimethylsulfoxide (DMSO), and toluene were all obtained from Shanghai Chemical Reagent Company and used without further purification. [Rh(nbd)Cl]2 was the product of Acros Organics.
Measurements
The melting point (mp) was measured on an X-4 micro-melting point apparatus. Fourier transform infrared (FT-IR) spectra were measured on a Bruker Tensor 27 FT-IR spectrometer at room temperature using KBr pellets. The spectra were obtained at a 4 cm−1 resolution and recorded in the region of 4000–400 cm−1. 1H NMR and 13C NMR spectra measurements were recorded on a Bruker AVANCE 300 NMR spectrometer using chloroform-d or DMSO-d6 as solvent and tetramethylsilane (TMS) as internal standard. Chemical shifts were recorded in ppm. The number-average molecular weight (Mn), weight-average molecular weight (Mw), and molecular weight polydispersities were determined by gel permeation chromatography (GPC) using THF as the eluent, calibrated by polystyrenes standards. The optical rotation was measured in a WZZ-2S (2SS) digital automatic polarimeter at room temperature. UV–vis absorption spectra in solutions were measured on a Shimadzu UV 3600 spectrometer. Circular dichroism (CD) spectra were determined with a Jasco J-810 spectropolarimeter at room temperature. Thermal analysis experiments were performed using a thermogravimetric analysis (TGA) apparatus operated in the conventional TGA mode (TA Q-600, TA Instruments) at the heating rate of 10 K/min in a nitrogen atmosphere. Infrared emissivity values of the samples were investigated on the IRE-I Infrared emissivity measurement instrument of Shanghai Institute of Technology and Physics, China.
Synthesis of l,d and dl-tyrosine methyl ester
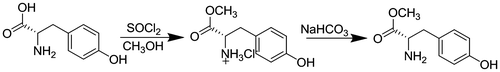
To 50 ml anhydrous methanol, 2.5 mL (30 mmol) thionyl chloride was added dropwise, with stirring, over a period of 15 min at 0 °C. To this mixture 5 g (27 mmol) l-tyrosine was added. The reaction mixture was slowly heated to 60 °C and refluxed for 6 h. After the reaction was finished, the superfluous thionyl chloride and methanol were evaporated on the rotary evaporator to get the l-tyrosine methyl ester hydrochloride.
l-tyrosine methyl ester hydrochloride (5 g) was dissolved in a saturated sodium bicarbonate solution (150 mL) and extracted with ethyl acetate (5 × 50 mL). After the resulting organic solution was concentrated under reduced pressure, the pure ester was obtained in a 95% yield. d- and dl-tyrosine methyl ester were synthesized in the similar way using d- and dl-tyrosine instead of l-tyrosine.
l-tyrosine methyl ester: a white solid, mp 132 °C, = +26.8° (c = 1 g/dL, methanol). 1H NMR (300 MHz, CDCl3): δ 7.04 (d, J = 6.9 Hz, 2H, phenyl), 6.73 (d, J = 5.5 Hz, 2H, phenyl), 3.75 (s, 4H, C*HCO2CH3), 3.05 (d, J = 13.1 Hz, 1H, CH2-Ph), 2.84 (d, J = 13.6 Hz, 1H, CH2-Ph). FT-IR (ν/cm−1): 3357, 3300 (–NH2), 1744 (C=O), 1601, 1513, 1478, 1442.
d-tyrosine methyl ester: a white solid, mp 134 °C, = −25.6° (c = 1 g/dL, methanol). Its IR and 1H NMR spectra are resembled to l-tyrosine methyl ester and thus omitted.
dl-tyrosine methyl ester: a white solid, mp 135 °C, = 0° (c = 1 g/dL, methanol). Its IR and 1H NMR spectra are resembled to l-tyrosine methyl ester and thus omitted.
Monomer synthesis
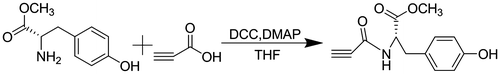
l-tyrosine methyl ester (0.98 g, 5 mmol), propiolic acid (0.35 g, 5 mmol), and DMAP (60 mg, 0.5 mmol) were added to THF (40 mL) sequentially. The solution was stirred at room temperature for 30 min, and then, DCC (1.24 g, 6 mmol) was added to the solution. After 12 h, white precipitate was formed and then filtered off, and the filtrate was collected, to which ethyl acetate (100 mL) was added to extract the desired product. The combined solution was washed with 2 M HCl and then washed with saturated aqueous NaHCO3 to neutralize the solution. Then, the solution was dried over anhydrous MgSO4, filtered, and concentrated to get the target monomer. The crude monomer was purified by flash column chromatography on silica gel (hexane/ethyl acetate) 1/1, v/v). A colorless solid was obtained in 69% yield. N-propioloyl-d-tyrosine methyl ester and N-propioloyl-dl-tyrosine methyl ester were synthesized in the similar way using d- and dl-tyrosine instead of l-tyrosine. The monomers were record as LM, DM, and RM which represented N-propioloyl-l-tyrosine methyl ester, N-propioloyl-d-tyrosine methyl ester and N-propioloyl-dl-tyrosine methyl ester.
The spectroscopic data of monomers are given below.
N-propioloyl-l-tyrosine methyl ester: 1H NMR (300 MHz, CDCl3): δ 6.94 (d, J = 7.2 Hz, 2H, phenyl), 6.78 (d, J = 7.6 Hz, 2H, phenyl), 6.63 (d, 1H, NH), 4.90 (m, 1H, NHCH), 3.77 (s, 3H, CO2CH3), 3.09 (m, 2H, CH2-Ph), 2.86 (s, 1H, ≡CH). 13C NMR (75 MHz, DMSO-d6): δ 171.73 (≡CCO2), 156.22 (C–OH), 151.87 (CO2CH3), 130.39, 127.64, 115.54, 77.90 (≡C), 77.20 (≡CH), 54.23, 52.41, 35.33. FT-IR (ν/cm−1): 3306 (≡CH), 3260 (N–H), 2108 (C≡C), 1743 (CO2CH3), 1645 (CONH).
N-propioloyl-d-tyrosine methyl ester: 1H NMR (300 MHz, CDCl3): δ 6.94 (d, J = 7.2 Hz, 2H, phenyl), 6.78 (d, J = 7.6 Hz, 2H, phenyl), 6.63 (d, 1H, NH), 4.90 (m, 1H, NHCH), 3.77 (s, 3H, CO2CH3), 3.09 (m, 2H, CH2-Ph), 2.86 (s, 1H, ≡CH). 13C NMR (75 MHz, DMSO-d6): δ 171.73 (≡CCO2), 156.22 (C–OH), 151.87(CO2CH3), 130.39, 127.64, 115.54, 77.90 (≡C), 77.20 (≡CH), 54.23, 52.41, 35.33. FT-IR (ν/cm−1): 3306 (≡CH), 3260 (N–H), 2108 (C≡C), 1743 (CO2CH3), 1645 (CONH).
N-propioloyl-dl-tyrosine methyl ester: 1H NMR (300 MHz, CDCl3): δ 6.94 (d, J = 7.2 Hz, 2H, phenyl), 6.78 (d, J = 7.6 Hz, 2H, phenyl), 6.63 (d, 1H, NH), 4.90 (m, 1H, NHCH), 3.77 (s, 3H, CO2CH3), 3.09 (m, 2H, CH2-Ph), 2.86 (s, 1H, ≡CH). 13C NMR (75 MHz, DMSO-d6): δ 171.73 (≡CCO2), 156.22 (C–OH), 151.87 (CO2CH3), 130.39, 127.64, 115.54, 77.90 (≡C), 77.20 (≡CH), 54.23, 52.41, 35.33. FT-IR (ν/cm−1): 3306 (≡CH), 3260 (N–H), 2108 (C≡C), 1743 (CO2CH3), 1645 (CONH).
Polymerization
The monomer was polymerized in the distilled THF solution using [Rh(nbd)Cl]2 as the catalyst. N-propioloyl-l-tyrosine methyl ester (0.247 g, 1 mmol) was added to a three-necked flask. The flask was evacuated and flushed with nitrogen three times. And then one drop of TEA and 5 mL dry THF were added. [Rh(nbd)Cl]2 (4.7 mg, 10 μmol) was added to another flask, dissolved in the solution of THF, and also placed under nitrogen atmosphere. The [Rh(nbd)Cl]2 solution was transferred to the N-propioloyl-l-tyrosine methyl ester solution, and the polymerization mixture was vigorously stirred under nitrogen for 24 h. After the reaction, the solution was concentrated and purified three times by reprecipitation with a large amount of n-hexane. The precipitate was filtered and dried under reduced pressure at room temperature to a constant weight. The other polymers were prepared in the similar way using DM, RM instead of LM. The polymers were record as LPA, DPA, and RPA, respectively.
The spectroscopic data of polymers are given below.
Poly(N-propioloyl-l-tyrosine methyl ester) (LPA): 1H NMR (300 MHz, DMSO-d6): δ 9.26 (NH), 7.02 (phenyl), 6.67 (phenyl), 5.76 (=CH), 4.39 (OH), 4.21 (NHCH), 3.61 (CO2CH3), 2.92 (CH2-Ph), 2.78 (CH2-Ph). FT-IR (ν/cm−1): 3268 (N–H),1738 (CO2CH3), 1647 (CONH).
Poly(N-propioloyl-d-tyrosine methyl ester) (DPA): 1H NMR (300 MHz, DMSO-d6): δ 9.26 (NH), 7.02 (phenyl), 6.67 (phenyl), 5.76 (=CH), 4.39 (OH), 4.21 (NHCH), 3.61 (CO2CH3), 2.92 (CH2-Ph), 2.78 (CH2-Ph). FT-IR (ν/cm−1): 3268 (N–H),1738 (CO2CH3), 1647 (CONH).
Poly(N-propioloyl-d-tyrosine methyl ester) (RPA): 1H NMR (300 MHz, DMSO-d6): δ 9.26 (NH), 7.02 (phenyl), 6.67 (phenyl), 5.76 (=CH), 4.39 (OH), 4.21 (NHCH), 3.61 (CO2CH3), 2.92 (CH2-Ph), 2.78 (CH2-Ph). FT-IR (ν/cm−1): 3268 (N–H),1738 (CO2CH3), 1647 (CONH).
Results and discussion
Monomer synthesis
The synthesis of monomers was conducted according to the reaction steps in Schemes and . Tyrosine methyl ester hydrochlorides were synthesized by reacting tyrosine with anhydrous methanol in the presence of thionyl chloride. Then, tyrosine methyl ester hydrochlorides were dissolved in a saturated sodium bicarbonate solution and extracted with ethyl acetate. After the resulting organic solution was concentrated under reduced pressure on the rotary evaporator, the tyrosine methyl esters were obtained. Then, the N-propioloyl-tyrosine methyl esters were prepared by the reaction of propiolic acid with tyrosine methyl esters in the presence of DCC and DMAP in freshly distilled THF. The structure of N-propioloyl-tyrosine methyl esters was confirmed by IR and 1H NMR and 13C NMR spectroscopy. As shown in Figure , the monomer exhibited a strong absorption bands at 2108 and 3306 cm−1 relative to the C≡C and ≡C–OH stretching vibrations, respectively, while these peaks were not absent in the IR spectrum of l-tyrosine methyl ester, from which the formation of N-propioloyl-l-tyrosine methyl ester was supposed. Figure illustrated the 1H NMR spectra of l-tyrosine methyl ester and the corresponding acetylene monomer in CDCl3. In the 1H NMR spectra of l-tyrosine methyl ester (Figure (a)) and N-propioloyl-l-tyrosine methyl ester (Figure (b)) measured in chloroform-d, there were no resonance peaks relative to the primary amine group and hydroxyl group, which was probably due to the chemical activity of hydrogen bonds in the structure of molecular. The signals assignable to the ethynyl proton (HC≡) around δ 2.8 ppm and amide proton (NH) at δ 6.6 ppm were appeared after the reaction compared with l-tyrosine methyl ester; thus, it could be conformed that N-propioloyl-l-tyrosine methyl ester was successfully prepared. Furthermore, 13C NMR spectrum of the monomer was supported to confirm the structure of N-propioloyl-l-tyrosine methyl ester. As shown in Figure , the acetylenic carbon atoms of N-propioloyl-l-tyrosine methyl ester resonated at δ 77.90 and 77.20, the absorption peaks of the ester carbonyl carbon and the carbonyl carbon linking to the triple bond were appeared at 171.73 and 151.87, respectively. The same results could be obtained instead l-tyrosine with d- and dl-tyrosine.
Figure 1. IR spectra of l-tyrosine methyl ester (a) and N-propioloyl-l-tyrosine methyl ester (b).
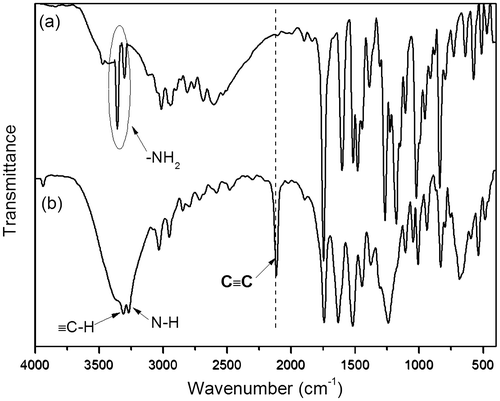
Figure 2. 1H NMR spectra in CDCl3 solutions of l-tyrosine methyl ester (a) and N-propioloyl-l-tyrosine methyl ester (b). The resonance peaks of the residual nondeuterated solvents, water dissolved in the deuterated solvents, and TMS are marked with asterisks.
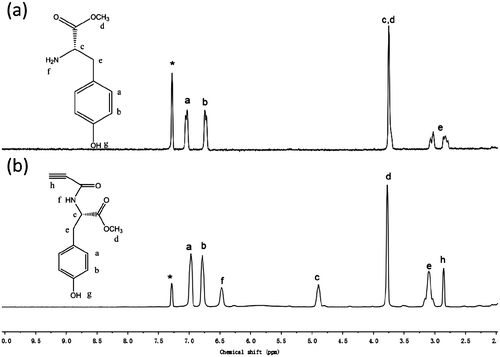
Figure 3. 13C NMR spectra in DMSO-d6 of N-propioloyl-l-tyrosine methyl ester. The resonance peaks of solvents are marked with asterisks.
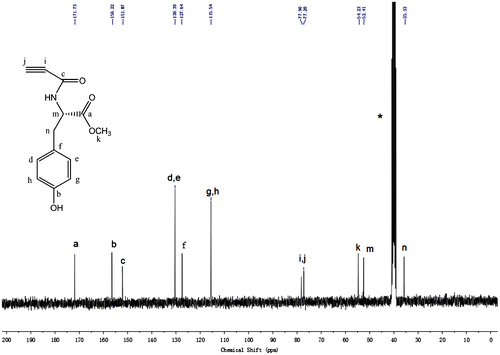
Figure 4. IR spectra of N-propioloyl-l-tyrosine methyl ester (a) and poly(N-propioloyl-l-tyrosine methyl ester) (b).
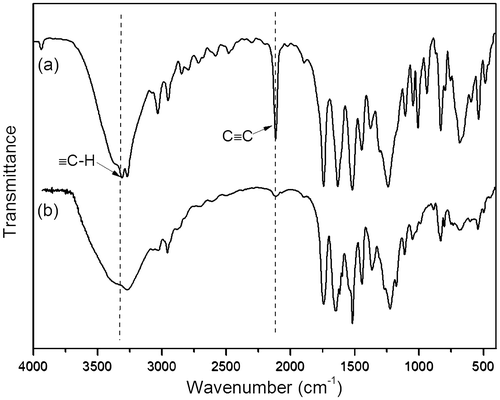
Figure 5. 1H NMR spectra in DMSO-d6 of poly(N-propioloyl-l-tyrosine methyl ester). The resonance peaks of the residual nondeuterated solvents are marked with asterisks.
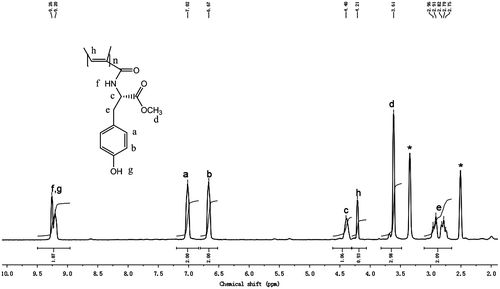
Figure 6. 1H NMR spectra of CDCl3 solutions of LM with concentrations of (a) 10, (b) 25, (c) 50, (d) 75, and (e) 100 mg/mL. The resonance peaks of the amide proton are marked with downward arrows, while those of solvent are marked with asterisks.
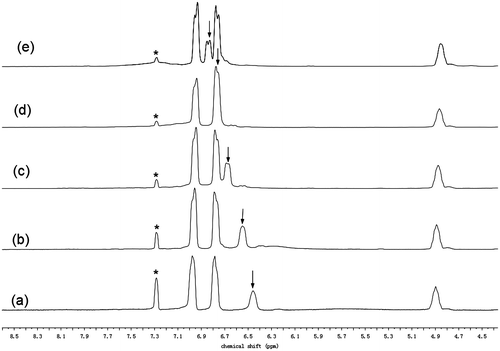
Figure 7. Concentration dependence of chemical shift of amide proton resonance of the CDCl3 solutions of N-propioloyl-l-tyrosine methyl ester.
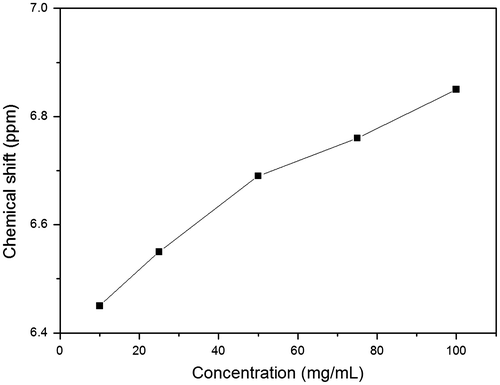
Figure 8. IR spectra of CHCl3 solutions of N-propioloyl-l-tyrosine methyl ester with concentrations of (a) 200, (b) 150,(c) 100, (d) 80, (e) 60, (f) 40, (g) 20, (h) 10, and (i) 1 mg/mL.
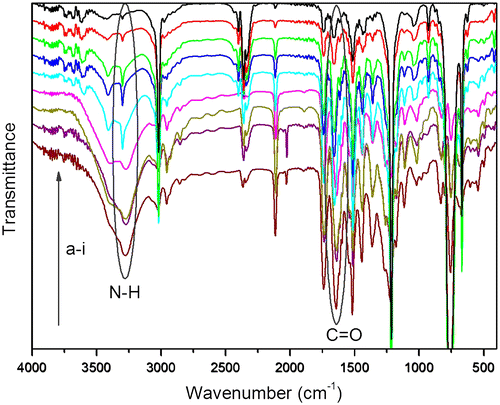
Figure 9. IR spectra of PAs at (a) 3600–3000 cm−1 and (b) 1800–1500 cm−1.
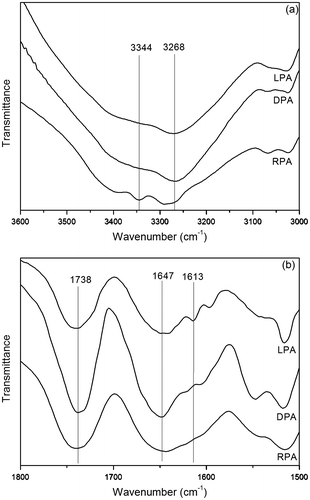
Figure 10. (a) CD spectra of PAs in THF (c = 0.01 g/dL) at room temperature. (b) UV–Vis spectra of PAs in THF (c = 0.01 g/dL) at room temperature.
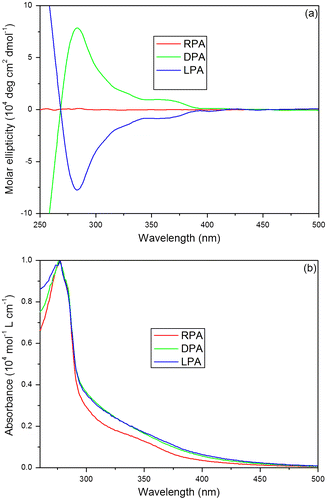
Figure 11. TGA thermograms of PAs recorded under a nitrogen atmosphere at a heating rate of 10 °C/min.
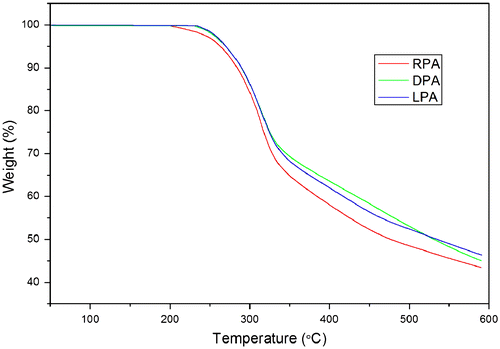
Figure 12. Infrared emissivity values of PAs measured at 25–150 °C.
Scheme 1. Synthesis of l-tyrosine methyl ester.
Scheme 2. Synthesis of N-propioloyl-l-tyrosine methyl ester.
Polymer synthesis
Polymerization of the monomers was carried out in THF using the rhodium catalyst [Rh(nbd)Cl]2 under nitrogen atmosphere (Scheme ). The progress of polymerization was confirmed by 1H NMR and IR spectroscopies. The results of the polymerization of N-propioloyl-tyrosine methyl esters are summarized in Table , while the number-average molecular weight (Mn) and weight-average molecular weight (Mw) are shown in Figure S1(a)–(c) and S2.
Scheme 3. Polymerization of N-propioloyl-l-tyrosine methyl ester.
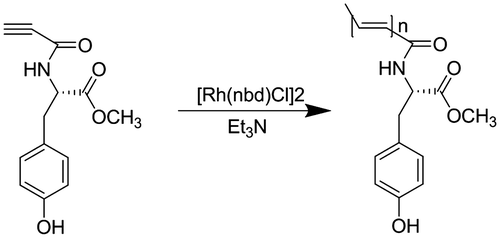
Table 1. Polymerization results of PAsTable Footnotea.
The structure of the polymer is analyzed by spectroscopic methods. The IR spectra of LPA and its corresponding monomer are shown in Figure . While the monomer exhibits strong absorption bands at 3266 and 2108 cm−1, due to the ≡C–H and C≡C stretching vibrations, respectively. These peaks completely disappear in the IR spectrum of the polymer, suggesting that the acetylene triple bond of N-propioloyl-l-tyrosine methyl ester has been consumed by the Rh-catalyzed polymerization.
According to the solubility of the polymers that the polyacetylenes have better solubility in DMSO-d6 than in CDCl3, we thus choose the 1H NMR spectra of polyacetylenes in DMSO-d6. Figure shows the 1H NMR spectra of LPA in the solution of DMSO-d6. As can be seen from the spectrum of polymer, the resonance peak is observed at δ 4.4 ppm, which is relative to the olefinic proton of the polymer. The resonance of olefinic proton, however, is not appeared in the spectrum of monomer. The spectral data of IR as well as 1H NMR indicates that the polymerization reaction has successfully converted the acetylenic triple bond in monomer to the olefinic double bond in polymer.
Hydrogen bond analysis
The 1H NMR spectrum is a useful method to investigate which kind of hydrogen bond is formed.[Citation39–43] When a proton is involved in hydrogen bond, its electrons are shared by two electronegative elements and its electron density is hence decreased. Therefore, the proton is deshielded and comes into resonance at a lower field.[Citation44–46] For intermolecular hydrogen bonds, the extent of hydrogen bond is affected by concentration. In the nonpolar solvent, the degree of hydrogen bond increases with the increase in the solution concentration, resulting in a downfield shift in the resonance peak of the proton.[Citation44–46] In the previous study, it has been found that both the poly(phenylacetylenes)s carrying l-leucine pendants and the poly(phenylacetylenes)s bearing l-valine pendants readily form intermolecular hydrogen bonds.[Citation6,47] Therefore, the formation of hydrogen bond of N-propioloyl-l-tyrosine methyl ester is also investigated by 1H NMR spectra in CDCl3 solutions with different concentrations in Figure . In the aprotic solvent, the position of the resonance peak of the amide proton changes with the change of the concentration. N-propioloyl-l-tyrosine methyl ester shows a resonance peak of amide proton at δ 6.45 in a dilute solution with concentration of 10 mg/mL (Figure (a)). When the concentration is increased to 25 mg/mL, a similar 1H NMR spectrum is observed; however, the resonance peak of amide proton is shifted to δ 6.55 (Figure (b)). Then, with the solution concentration increasing gradually, the amide resonance peak is still progressively downfield shifted. When the solution concentration is increased to 100 mg/mL, the amide resonance peak is downfield shifted to δ 6.85 (Figure (e)). Obviously, the downfield shift of the amide proton resonance peak accompanying the solution concentration increasing must be due to the formation of intermolecular hydrogen bonds. Plotting the NMR data in Figure indicates that the resonance peak position of amide proton downfield shifts with the increase in the solution concentration. This evidence reveals that the degree of hydrogen bond increases with the solution concentration. And the deshielding effect should be caused by the self-association of N-propioloyl-l-tyrosine methyl ester molecules via the formation of multiple hydrogen bonds between the N–H group of one molecule and the C=O group of another, which results in the downfield shift of the resonance peak. Then, it can be inferred that the tyrosine pendants in the polymer are covalently bind to every repeat unit of the polyacetylene chains, the amide groups which locates closely can easily form hydrogen bond in the same or different polymer chains.
The IR spectrum is not only useful in the analysis the structure of molecules; it is also an important method for the analysis of hydrogen bond. In order to confirm which kind of hydrogen bond is formed in the molecules, the IR spectrum of LPA is conducted in CHCl3 with different concentrations.[Citation8,10] As shown in Figure (a)–(i), with the concentrations of LPA in CDCl3 reducing from 300 to 10 mg/mL, the peaks of hydrogen bonded N–H groups reduce remarkably along with the enhancement of free N–H groups. Moreover, the peaks of N–H groups become sharper. This evidence demonstrates that the most extent of hydrogen bonds in the LPA is intermolecular hydrogen bond. In addition, there are almost no differences when the concentrations of LPA are reduced to below 10 mg/mL, and hydrogen bond is still existed in the molecules even at a very low concentration, which demonstrates the existence of intramolecular hydrogen bond in the molecules.
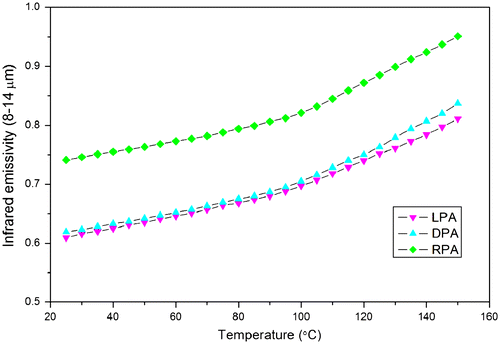
Participation in hydrogen bond could decrease the frequency of the C=O and N–H vibration.[Citation48–50] As shown in Figure (a), there are some differences in the region of 3000–3600 cm−1 corresponding to N–H stretching vibration of PAs. For RPA as seen in Figure (a), the bands at 3344 and 3268 cm−1 may be assigned to the free N–H groups and the hydrogen bonded N–H groups, respectively. For LPA and DPA, the band at 3344 cm−1 belonging to the free N–H groups has disappeared and the intensity of the absorption at 3268 cm−1 corresponds to the hydrogen bonded N–H groups have increased. This result indicates that the free N–H groups have converted to be the hydrogen bonded N–H groups, and the LPA and DPA possess a higher degree of hydrogen bond than the RPA. The FT-IR spectra of PAs in the carbonyl stretching regions are shown in Figure (b). As can be seen, there are two peaks, centered at 1738 and 1647 cm−1, associated with the carbonyl groups belonging to esters and amides, respectively. Meanwhile, a hydrogen bonded carbonyl band could be observed at a lower frequency of 1613 cm−1 in LPA and DPA, it is, however, weak and almost cannot be observed in the RPA. Compare with the ester carbonyl absorption, the amide carbonyl absorption decreases as the hydrogen bonded carbonyl absorption increases in the spectra of LPA and DPA, which also indicates that the N–H groups are involved in hydrogen bond with carbonyl groups in the amide, and the LPA and DPA possess a higher degree of hydrogen bond than the RPA. From the IR and NMR analyses, both intra- and intermolecular hydrogen bonds are existed in the LPA and DPA, and their unique properties would be investigated in the next.
Optical rotation and chain helicity
The chain conformations of PAs were investigated by polarimetry, CD, and UV–vis spectroscopies. A polymer chain carrying chiral pendants can possibly show very high optical activity [Citation9,19,51,52]; thus, the specific optical rotations of our polymers and those of their corresponding monomers were measured to study whether they are optically active. The specific optical rotations of all the polymers and monomers are summarized in Table . LPA and DPA exhibit high
values, suggesting that they are highly optically active, while RPA has no optical activity. The
values of LPA and DPA are much higher than those of their corresponding monomers in the same solvents. For example, the
value for the THF solution of LPA (+154.2°) is 10-fold higher than that for LM (+15.0°). The large specific rotation difference between the monomer and the polymer indicates that the main chains of the polymer take the helical conformations. As is known to all that a polymer chain with a helical conformation can exhibit very high optical activity.[Citation9,19,37,51,52] Then, the high
values of LPA and DPA demonstrate that the large specific rotation difference between the monomer and the polymer may be result from the helical secondary structure. In other words, the polymer chains have been induced by the asymmetric forces of the chiral l-tyrosine and d-tyrosine pendants groups to take a helical conformation.
Table 2. Specific optical rotation of the chiral monomers and their helical polymers in different solventsTable Footnotea.
The high specific optical rotations values of LPA and DPA suggest that their chain segments may possess the helical conformations, while RPA is optically in-active. To further verify this, we analyzed with CD spectroscopy, which is a significant method for the research of secondary structure. Figure (a) shows the CD spectra of polyacetylenes in THF at room temperature. While RPA is CD in-active at wavelengths longer than 260 nm, LPA and DPA exhibit strong Cotton effects with excellent chiral symmetry at ~285 and ~365 nm in the long wavelength region, which are relative to the absorptions of the segments of polyacetylene backbone. This undoubtedly proves that the main chain of LPA and DPA possess the left-handed and right-handed helical conformation. Then, the structure of LPA and DPA is more orderly than that of RPA. That also exhibits that the degree of hydrogen bonds of LPA and DPA are higher than that of RPA according to the hydrogen bond analysis. Hence, we can come to a conclusion that the helical conformation of the polyacetylene chain could be induced by the pendant chirality. The asymmetric force field generated by the chiral pendants may drive the polymer chain to rotate in a screw sense.
The UV absorption spectra of polyacetylenes measured in THF at room temperature are depicted in Figure (b). As can be seen in Figure (b), the UV absorption spectra of polyacetylenes are almost the same. All three polymers show not only a strong absorption at ~280 nm belong to the n–π* electronic transition of the carbonyl group but also strong absorptions in the long wavelength region because of the electronic absorption of the polyacetylene backbone. Furthermore, the polyene backbone absorption of RPA is, however, a little weaker than that of LPA and DPA. The low absorptivity of RPA may be due to the lack of secondary structure in the main chain, which reduces the effective conjugation length. Some research groups have found that the absorption of a polyacetylene chain increases with its stereoregularity.[Citation53] Hence, we can conclude that LPA and DPA have more orderly secondary structure and than RPA.
Thermal stability and solubility
The thermal stability of PAs was investigated by thermogravimertric analysis at a heating rate of 10 °C/min under a nitrogen atmosphere. As shown in Figure , the TGA curve for each polymer exhibits a smooth thermal degradation. LPA and DPA are thermally very stable and virtually do not lose any weight when they are heated to temperature as high as ~231 °C, whereas RPA starts to lose its weight at ~201 °C. The decomposition temperatures of PAs are given in Table . The 5 and 10% weight loss temperatures (T5, T10) of the polymers have been taken as standard to evaluate the thermal stability of the polymers. Monosubstituted polyacetylene such as poly(1-hexyne) is thermally unstable relatively, which starts to lose its weight at ~150 °C.[Citation54] The enhanced thermal stability of PAs is probably due, at least partly, to the electronic effect of the pendant groups. The electron-withdrawing carbonyl group could reduce the reactivity of the polymer and form resonance structure through its electronic interactions with the polyene backbone to stabilize or deactivate the radical species and hence enhance the resistance of the polymers against thermal degradation. Moreover, neither LPA nor DPA, however, loses any weight below 230 °C in the TGA. Optically active LPA and DPA are thermally more stable than the racemic RPA without helical conformation. The thermally stable helical polyacetylenes have more regulated secondary structure, which could form a high degree of intra- and interchain hydrogen bonds in the structure. Hydrogen bond interaction could rigidify the polymer chains, thus enhancing their resistance to thermolysis. Consequently, LPA and DPA have better ability to protect the polymer backbone from the thermal attack, which benefit from the helical conformation.
Table 3. Thermal decomposition temperature of PAs.
As shown in Table , all of our three polymers show the similar solubility, which are completely soluble in common organic solvents such as methanol, THF, DMF, and DMSO, partially soluble in dichloromethane, chloroform, and insoluble in toluene. The reason for this solubility phenomenon may be due to the helical structure and intra- and interchain hydrogen bonds in the polymers, which can interact with the polar solvents. Consequently, polyacetylenes have better resolution in polar solvents than nonpolar solvents.
Table 4. Solubility of PAs in different solventsTable Footnotea.
Infrared emissivity analysis
As can be seen from the TGA thermograms of PAs, no weight begins to lose when heated to 200 °C. The infrared emissivity values of PAs at wavelength of 8–14 μm are measured from 25 to 150 °C. As shown in Table , the polymer carrying racemic tyrosine has a higher infrared emissivity value of 0.741 at room temperature (25 °C), while LPA and DPA with the helical conformations possess lower values of 0.611 and 0.632, respectively. Undoubtedly, the reduction in infrared emissivity value is relative to the more ordered stereo structure and secondary structure derived from the chiral tyrosine pendants. Figure exhibits that the infrared emissivity values of PAs are increased with the temperature. The thermal energy derived from the vibration of molecules induced the infrared radiation.[Citation6,55] Therefore, the thermal controlling of molecular vibration could be favored to reduce the infrared emissivity values. LPA and DPA with the more ordered stereo structure and secondary structure could be easier to form a high degree of intra- and interchain hydrogen bonds and reduce thermal energy of molecular vibration, thus reducing the infrared emissivity values.
Table 5. Infrared emissivity values of PAs at room temperature.
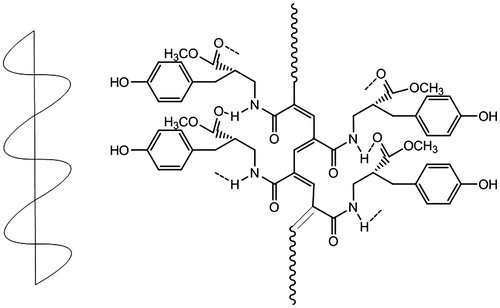
It is obvious that the unsaturated groups (–NHCO– in PAs) in the structure of polymer chain induce the high infrared emissivity values of the polymer. According to the 1H NMR and FT-IR spectra analysis, PAs exhibit a high degree of intra- and interchain hydrogen bonds between C=O and –NH groups (Scheme ). LPA and DPA with helical conformation could be easy to form hydrogen bonds among the high unsaturated groups; thus, the degree of hydrogen bonds in optically active polyacetylene is definitely higher than that in racemic one. It is well known that the polymer with a higher degree of hydrogen bonds in the structure could reduce the index of hydrogen deficiency and the unsaturated degree.[Citation6,55] Thus, the infrared emissivity values of LPA and DPA are lower than that of RPA.
Scheme 4. Intrachain hydrogen bonds interaction.
Conclusions
In this work, N-propioloyl-tyrosine methyl ester carrying chiral and racemic tyrosine pendants were synthesized and then polymerized in the presence of [Rh(nbd)Cl]2 as the catalyst, providing the corresponding optically active and racemic polyacetylenes (LPA, DPA and RPA) in high yields. LPA and RPA take the helical conformations, exhibiting strong Cotton effect due to the absorptions of their polyacetylene backbone in the CD spectra. The LPA and DPA are a couple of enantiomers, the specific optical rotations of them are reached to +154.2° and −158.2° in the THF, respectively, which larger than their corresponding monomers containing the chiral pendants, manifesting the influence of chiral amino acid pendants on the helical conformations of the polymer chains. The asymmetric force field in the chiral pendants induces stereoregularity in the polymer chain. Unlike monosubstituted polyacetylenes such as poly(1-hexyne), PAs especially the optically active polyacetylenes are thermally stable. The enhanced thermal stability of PAs is probably relative to the electronic effects of the pendant groups, helical conformations, and higher degree of hydrogen bonds of the polymer chains. The LPA and DPA possess the helical conformations and higher degree of hydrogen bonds compared to the RPA, which have random coiled polymer chain. To some extent, the helical structure of polyacetylene plays essential roles for the polymer to form hydrogen bonds in the structure. Thus, the optically active polyacetylenes carrying chiral amino acid pendants could be taken as a kind of promising materials with low infrared emissivity in the field of stealthy technology. It proposes a possibility to adjust the infrared emissivity value with the change of the polymer structure.
Supplemental data
Supplemental data for this article can be accessed here. http://dx.doi.10.1080/15685551.2014.907620
TDMP_A_907620_Suppl.doc
Download MS Word (1.5 MB)Acknowledgements
The authors are grateful to the National Nature Science Foundation of China (51077013), Fund Project for Transformation of Scientific and Technological Achievements of Jinagsu Province of China (BA2011086) for financial support.
References
- Mahulikar SP, Sonawane HR, Arvind Rao G. Infrared signature studies of aerospace vehicles. Prog. Aerosp. Sci. 2007;43:218–245.10.1016/j.paerosci.2007.06.002
- Huang Z, Zhu D, Lou F, Zhou W. An application of Au thin-film emissivity barrier on Ni alloy. Appl. Surf. Sci. 2008;255:2619–2622.10.1016/j.apsusc.2008.07.185
- Yu H, Xu G, Shen X, Yan X, Cheng C. Low infrared emissivity of polyurethane/Cu composite coatings. Appl. Surf. Sci. 2009;255:6077–6081.10.1016/j.apsusc.2009.01.019
- Wang Z, Zhou Y, Sun Y. Helical polyurethane-imide@attapulgite composite: preparation, characterization and infrared emissivity study. Mater. Lett. 2010;64:908–911.10.1016/j.matlet.2010.01.055
- Ye X, Zhou Y, Chen J, Sun Y, Wang Z. Coating of ZnO nanorods with nanosized silver particles by electroless plating process. Mater. Lett. 2008;62:666–669.10.1016/j.matlet.2007.06.037
- Wang Z, Zhou Y, Sun Y, Yao Q. Optically active helical polyurethane−urea with single-handed conformation for infrared low emissivity. Macromolecules. 2009;42:4972–4976.
- Liu J, Lam JW, Tang BZ. Acetylenic polymers: syntheses, structures, and functions. Chem. Rev. 2009;109:5799–5867.10.1021/cr900149d
- Nomura R, Tabei J, Masuda T. Biomimetic stabilization of helical structure in a synthetic polymer by means of intramolecular hydrogen bonds. J. Am. Chem. Soc. 2001;123:8430–8431.10.1021/ja015688+
- Lam JW, Dong Y, Cheuk KK, Law CC, Lai LM, Tang BZ. Helical conjugated polymers: synthesis, stability, and chiroptical properties of poly(alkyl phenylpropiolate)s bearing stereogenic pendants. Macromolecules. 2004;37:6695–6704.10.1021/ma048960j
- Nomura R, Tabei J, Nishiura S, Masuda T. A helix in helices: a helical conjugated polymer that has helically arranged hydrogen-bond strands. Macromolecules. 2002;36:561–564.
- Feringa BL, van Delden RA, Koumura N, Geertsema EM. Chiroptical molecular switches. Chem. Rev. 2000;100:1789–1816.10.1021/cr9900228
- Nakano T, Okamoto Y. Synthetic helical polymers: conformation and function. Chem. Rev. 2001;101:4013–4038.10.1021/cr0000978
- Manesiotis P, Hall AJ, Emgenbroich M, Quaglia M, Lorenzi ED, Sellergren B. An enantioselective imprinted receptor for Z-glutamate exhibiting a binding induced color change. Chem. Commun. 2004;2278–2279.10.1039/b407870e
- Buryak A, Severin K. A chemosensor array for the colorimetric identification of 20 natural amino acids. J. Am. Chem. Soc. 2005;127:3700–3701.10.1021/ja042363v
- Nishiyabu R, Anzenbacher P. Sensing of antipyretic carboxylates by simple chromogenic calix[4]pyrroles. J. Am. Chem. Soc. 2005;127:8270–8271.10.1021/ja051421p
- Yashima E, Maeda K, Iida H, Furusho Y, Nagai K. Helical polymers: synthesis, structures, and functions. Chem. Rev. 2009;109:6102–6211.10.1021/cr900162q
- Nakai T, AbdulKarim S, Teraguchi M, Sanda F, Masuda T. Synthesis and properties of polyacetylenes having substituted azobenzene pendant groups. 2002;39:901–913.
- Rahim EA, Sanda F, Masuda T. Synthesis and properties of a novel polyacetylene containing eugenol moieties. J. Macromol. Sci. Part A Pure Appl. Chem. 2004;41:133–141.10.1081/MA-120027299
- Lam JW, Dong Y, Cheuk KK, Tang BZ. Helical disubstituted polyacetylenes: synthesis and chiroptical properties of poly(phenylpropiolate)s. Macromolecules. 2003;36:7927–7938.10.1021/ma0349433
- Onouchi H, Hasegawa T, Kashiwagi D, Ishiguro H, Maeda K, Yashima E. Helicity induction in charged poly(phenylacetylene)s bearing various acidic functional groups in water and its mechanism. Macromolecules. 2005;38:8625–8633.10.1021/ma051617+
- Tang BZ, Kotera N. Synthesis of optically active polyacetylene containing an asymmetric silicon by using organotransition-metal complexes as catalysts. Macromolecules. 1989;22:4388–4390.10.1021/ma00201a040
- Lam JWY, Dong Y, Cheuk KKL, Luo J, Xie Z, Kwok HS, Mo Z, Tang BZ. Liquid crystalline and light emitting polyacetylenes: synthesis and properties of biphenyl-containing poly(1-alkynes) with different functional bridges and spacer lengths. Macromolecules. 2002;35:1229–1240.10.1021/ma011406e
- Lam JWY, Kong X, Dong Y, Cheuk KKL, Xu K, Tang BZ. Synthesis and properties of liquid crystalline polyacetylenes with different spacer lengths and bridge orientations. Macromolecules. 2000;33:5027–5040.10.1021/ma992097j
- Huang YM, Ge W, Lam JW, Tang BZ. Influence of electric field on the photoluminescence of a liquid crystalline monosubstituted polyacetylene. Appl. Phys. Lett. 2001;78:1652–1654.10.1063/1.1357806
- Huang YM, Lam JWY, Cheuk KKL, Ge W, Tang BZ. Monomer and dimer emissions in the solutions of a monosubstituted polyacetylene. Mater. Sci. Eng., B. 2001;85:118–121.10.1016/S0921-5107(01)00542-6
- Huang YM, Yip Lam JW, Leung Cheuk KK, Ge W, Tang BZ. Electrically tunable photoluminescence of liquid crystalline polyacetylene solutions. Mater. Sci. Eng., B. 2001;85:122–125.10.1016/S0921-5107(01)00543-8
- Yi H, Lujia B, Lewei B, Dezhen Z, Chengwei S, Zhongde X, Yip LW, Benzhong T, Jimmy MW. Characterization of novel liquid crystalline polymers of polyacetylenes having a comb-like structure via SEC/RI/RALLS/DV. Polym. Bull. 2000;44:539–546.10.1007/s002890070076
- Sun JZ, Chen HZ, Xu RS, Wang M, Lam JWY, Tang BZ. Electric field induced cis-to-trans isomerization of polyphenylacetylene in solid state. Chem. Commun. 2002;1222–1223.10.1039/b200830k
- Tang BZ, Chen HZ, Xu RS, Lam JWY, Cheuk KKL, Wong HNC, Wang M. Structure−property relationships for photoconduction in substituted polyacetylenes. Chem. Mater. 1999;12:213–221.
- Pui-Sze Lee P, Geng Y, Kwok HS, Tang BZ. Synthesis and light-emitting properties of poly(carbazolylacetylenes). Thin Solid Films. 2000;363:149–151.10.1016/S0040-6090(99)01029-9
- Tang B, Poon W, Peng H, Wong N, Ye X, Monde T. Synthesis and optical properties of poly (thienylacetylenes). Chin. J. Polym. Sci. 1999;17:81–86.
- Chen HZ, Xu RS, Sun Q, Lam JW, Wang M, Tang BZ. Photoconductivity of substituted polyacetylenes. Polym. Adv. Technol. 2000;11:442–449.10.1002/(ISSN)1099-1581
- Kong X, Tang BZ. Synthesis and novel mesomorphic properties of the side-chain liquid crystalline polyacetylenes containing phenyl benzoate mesogens with cyano and methoxy tails. Chem. Mater. 1998;10:3352–3363.10.1021/cm9803152
- Tang BZ, Kong X, Wan X, Peng H, Lam WY, Feng X-D, Kwok HS. Liquid crystalline polyacetylenes: synthesis and properties of poly{n-[((4′-cyano-4-biphenylyl)oxy)carbonyl]-1-alkynes}. Macromolecules. 1998;31:2419–2432.10.1021/ma971672l
- Lai LM, Lam JW, Qin A, Dong Y, Tang BZ. Synthesis, helicity, and chromism of optically active poly(phenylacetylene)s carrying different amino acid moieties and pendant terminal groups. J. Phys. Chem. B. 2006;110:11128–11138.10.1021/jp057179q
- Cheuk KK, Lam JW, Lai LM, Dong Y, Tang BZ. Syntheses, hydrogen-bonding interactions, tunable chain helicities, and cooperative supramolecular associations and dissociations of poly(phenylacetylene)s bearing l-valine pendants: toward the development of proteomimetic polyenes. Macromolecules. 2003;36:9752–9762.10.1021/ma035340u
- Cheuk KK, Li BS, Lam JW, Xie Y, Tang BZ. Synthesis, chain helicity, assembling structure, and biological compatibility of poly(phenylacetylene)s containing l-alanine moieties. Macromolecules. 2008;41:5997–6005.10.1021/ma800976e
- Li BS, Cheuk KK, Ling L, Chen J, Xiao X, Bai C, Tang BZ. Synthesis and hierarchical structures of amphiphilic polyphenylacetylenes carrying l-valine pendants. Macromolecules. 2003;36:77–85.10.1021/ma0213091
- Gemmecker G. Direct detection of hydrogen bonds in biopolymers by NMR spectroscopy. Angew. Chem. Int. Ed. 2000;39:1224–1226.10.1002/(SICI)1521-3773(20000403)39:7<1224::AID-ANIE1224>3.0.CO;2-X
- Zeng H, Miller RS, Flowers RA, Gong B. A highly stable, six-hydrogen-bonded molecular duplex. J. Am. Chem. Soc. 2000;122:2635–2644.10.1021/ja9942742
- Barchi JJ, Huang X, Appella DH, Christianson LA, Durell SR, Gellman SH. Solution conformations of helix-forming β-amino acid homooligomers. J. Am. Chem. Soc. 2000;122:2711–2718.10.1021/ja9930014
- Xu X-P, Au−Yeung SCF. Investigation of chemical shift and structure relationships in nucleic acids using NMR and density functional theory methods. J. Phys. Chem. B. 2000;104:5641–5650.
- Syud FA, Stanger HE, Gellman SH. Interstrand side chain−side chain interactions in a designed β-Hairpin: significance of both lateral and diagonal pairings. J. Am. Chem. Soc. 2001;123:8667–8677.10.1021/ja0109803
- Lambert JB, Shurvell HF, Lightner DA, Cooks RG. Organic structural spectroscopy. Upper Saddle River, NJ: Prentice Hall; 1998.
- Buhlmann P. Structure determination of organic compounds: tables of spectral data. Berlin: Springer; 2009.
- Silverstein R, Webster F. Spectrometric identification of organic compounds. New York, NY: Wiley; 2006.
- Cheuk KK, Lam JW, Chen J, Lai LM, Tang BZ. Amino acid-containing polyacetylenes: synthesis, hydrogen bonding, chirality transcription, and chain helicity of amphiphilic poly(phenylacetylene)s carrying l-leucine pendants. Macromolecules. 2003;36:5947–5959.10.1021/ma0344543
- Mattia J, Painter P. A comparison of hydrogen bonding and order in a polyurethane and poly(urethane−urea) and their blends with poly(ethylene glycol). Macromolecules. 2007;40:1546–1554.10.1021/ma0626362
- Yilgör E, Yurtsever E, Yilgör I. Hydrogen bonding and polyurethane morphology. II. Spectroscopic, thermal and crystallization behavior of polyether blends with 1,3-dimethylurea and a model urethane compound. Polymer. 2002;43:6561–6568.10.1016/S0032-3861(02)00566-9
- Coleman MM, Lee KH, Skrovanek DJ, Painter PC. Hydrogen bonding in polymers. 4. Infrared temperature studies of a simple polyurethane. Macromolecules. 1986;19:2149–2157.10.1021/ma00162a008
- Akagi K, Guo S, Mori T, Goh M, Piao G, Kyotani M. Synthesis of helical polyacetylene in chiral nematic liquid crystals using crown ether type binaphthyl derivatives as chiral dopants. J. Am. Chem. Soc. 2005;127:14647–14654.10.1021/ja051548e
- Fukushima T, Takachi K, Tsuchihara K. Optically active poly(phenylacetylene) film: simultaneous change of color and helical structure. Macromolecules. 2006;39:3103–3105.10.1021/ma0527597
- Tang BZ, Kong X, Wan X, Feng X-D. Synthesis and properties of stereoregular polyacetylenes containing cyano groups, poly[[4-[[[n-[(4′-cyano-4-biphenylyl)- oxy]alkyl]oxy]carbonyl]phenyl]acetylenes]. Macromolecules. 1997;30:5620–5628.10.1021/ma970409h
- Masuda T, Tang BZ, Higashimura T, Yamaoka H. Thermal degradation of polyacetylenes carrying substituents. Macromolecules. 1985;18:2369–2373.10.1021/ma00154a006
- Yang Y, Zhou Y, Ge J, Wang Y, Chen X. Synthesis, characterization and infrared emissivity property of optically active polyurethane derived from tyrosine. Polymer. 2011;52:3745–3751.10.1016/j.polymer.2011.06.027