
Abstract
The purpose of this research was to synthesize and characterize gelatin and tamarind gum (TG)/carboxymethyl tamarind (CMT) gum-based phase-separated hydrogels. The hydrogels were thoroughly characterized using bright-field microscope, FTIR spectroscope, differential scanning calorimeter, mechanical tester, and impedance analyzer. The mucoadhesivity, biocompatibility, and swelling property of the hydrogels were also evaluated. The antimicrobial efficiency of ciprofloxacin (model antimicrobial drug) loaded hydrogels was studied against E. coli. The in vitro drug release was carried out in both gastric and intestinal pHs. Microstructural analysis suggested the formation of phase-separated hydrogels. FTIR studies suggested that CMT gum altered the secondary structure of the gelatin molecules. Presence of the polysaccharides within the hydrogels resulted in the increase in the enthalpy and entropy for evaporation of the moisture from the hydrogels. The mechanical studies indicated viscoelastic nature of the hydrogels. Electrical analysis suggested an increase in the impedance of the hydrogels in the presence of the TG. The presence of CMT gum resulted in the decrease in the impedance of the hydrogels. The hydrogels exhibited good mucoadhesivity, biocompatibility, and pH-dependent swelling behavior. The drug-loaded hydrogels showed good antimicrobial activity and the drug release from the hydrogels was pH dependent and diffusion mediated.
1. Introduction
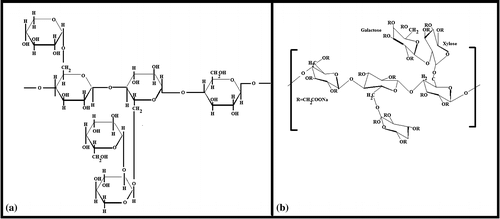
Polysaccharides are generally obtained from plant sources and are usually biocompatible.[Citation1] Due to their inherent biocompatibility, polysaccharides have been explored to design polymeric constructs of biomedical importance (pharmaceutical, cosmetic, and tissue engineering applications).[Citation2] The mechanical properties of the polysaccharide-based polymeric constructs are usually poor. Scientists have applied various methodologies to improve the mechanical properties of the polysaccharide constructs.[Citation3] Among the various methodologies, the commonly used techniques include blending the polysaccharides with other polymers (e.g. gelatin, PVA etc.) and crosslinking (chemical and physical) of the polymeric constructs.[Citation4] The commonly studied polysaccharides include starch, carboxymethyl cellulose, methyl cellulose, chitosan, alginate, carboxymethyl chitosan, and carboxymethyl starch. In recent years, tamarind gum (TG), due to its thickening property, has been explored as a natural polysaccharide for the development of pharmaceutical formulations [Citation5] and food products.[Citation6] The thickening property of TG helps stabilizing emulsions and induce gelation of the aqueous phase.[Citation7] TG is extracted from the seeds of the plant, Tamarindus indica.[Citation8] The backbone of TG consists of β-(1,4)-d-glucan substituted with side chains of α-(1,4)-d-xylopyranose and [Citation1,6] linked [β-d-galactopyranosyl-(1,2)-α-d-xylopyranosyl] to glucose residues.[Citation8,9] TG has been reported to be non-carcinogenic and non-toxic (biocompatible) in nature.[Citation10] The addition of TG improves the mucoadhesive property of the pharmaceutical formulations. The main disadvantages of TG include unpleasant odor and quick microbial degradation. To overcome these disadvantages, the derivatization of TG by chemical treatment has been explored. Carboxymethylation is one such chemical modification. Introduction of carboxymethyl group in TG makes the polymer anionic.[Citation11] This improves the hydration of the polysaccharide, thereby, resulting in the higher viscosity of the carboxymethylated product. It has been reported that the increase in the viscosity lowers the biodegradation of the polysaccharide.[Citation12] Though TG has been extensively studied for developing delivery vehicles for nutraceutical and pharmaceutical agents, carboxymethyl tamarind gum (CMT) has not been explored to that extent even though it holds a great promise in developing controlled release systems. The difference in the chemical structure of TG and CMT has been shown in Figure .
Figure 1. Chemical structures of (a) tamarind gum and (b) CMT gum.
As mentioned earlier, polysaccharides are seldom used alone for devising polymeric constructs. In this regard, gelatin/polysaccharide-based composite hydrogels have been well explored.[Citation13] Gelatin/polysaccharide-based hydrogels usually forms phase-separated hydrogels.[Citation14] This may be explained to the thermodynamic incompatibility of the polymer mixture during gelation. This, in turn, results in the formation of two phases: (a) polysaccharide rich phase and (b) gelatin rich phase. Since both the phases are aqueous in nature, the formed composite hydrogels are often regarded as water-in-water emulsions.[Citation15] The commonly used polysaccharides for developing such systems include (but not limited to) starch, soluble starch, hydrated starch, carboxymethyl starch, carboxymethyl cellulose dextran, and maltodextran. No reports were found to study the properties of gelatin-TG and gelatin-CMT phase-separated hydrogels.
Taking inspiration from the above, we have tried to develop gelatin-TG and gelatin-CMT-based phase-separated hydrogels. The physicochemical, thermal, and electrochemical properties of the hydrogels were thoroughly characterized using FTIR spectroscopy, differential scanning calorimetry, static mechanical tester, and impedance analyzer. The biological activity of the hydrogels was studied by mucoadhesive and biocompatibility (hemocompatibility and cell viability assay) studies. To understand the ability of the developed hydrogels as vehicles for controlled release, the hydrogels were loaded with ciprofloxacin (fluoroquinolone antibiotic). The drug release kinetics and the antimicrobial activity of the drug-loaded hydrogels were also studied in depth.
2. Materials and methods
2.1. Materials
TG and CMT (degree of carboxylation of CMT is 0.372) were procured from Maruti hydrocolloids, India. Gelatin was procured from Himedia, Mumbai, India. Ciprofloxacin was procured from Fluka Biochemical, China. Ethanol was obtained from Honyon International Inc., Hong Yang Chemical Corporation, China. Glutaraldehyde (25%, for synthesis; GA) and hydrochloric acid (35% pure) were obtained from Merck Specialities Private Limited Mumbai, India. Goat intestine and blood were collected from the local butcher shop. Double distilled water was used throughout the study.
2.2. Preparation of the formulations
Stock solution of gelatin (20% w/w) and polysaccharides (2% w/w) were freshly prepared. The stock solutions were maintained at 50 °C. The gelatin and polysaccharide solutions were mixed together (100 rpm, 10 min) in varying proportions (Table ) followed by addition of crosslinking reagent (0.5 ml of GA, 0.5 ml of ethanol and 0.01 ml of 0.1 N HCl). The mixture was mixed for 10 s and subsequently poured into petri plates/cylindrical molds. The petri plates/molds were incubated at room temperature (25 °C) for 1 h to induce gelation.
Table 1. Composition of the hydrogels.
Drug-loaded hydrogels were prepared by dispersing 0.1 g of ciprofloxacin in gelatin solution. Ciprofloxacin containing gelatin solution was used for the preparation of the hydrogels. Rest of the process remained same. The final concentration of the drug in the hydrogels was 0.5% w/w.
2.3. Microscopy studies
The microstructures of the uncrosslinked physical formulations were visualized under bright-field microscope (LEICA-DM 750 equipped with ICC 50-HD camera, Germany). The formulations were converted into thin smears over glass slides before visualization.
2.4. Infrared spectroscopy
The raw materials and the hydrogels were analyzed using FTIR spectrophotometer (Alpha-E, Bruker, USA). The analysis was done in the wavenumber range of 4500–450 cm−1. The spectrophotometer was being operated in the ATR mode.
2.5. Thermal analysis
The thermal profiles of the raw materials and the dried hydrogels were tested using differential scanning calorimeter (DSC 200 F3 Maia, Netzsch, Germany) in the temperature range of 40–400 °C under nitrogen atmosphere. The rate of thermal scanning was 5 °C/min.
2.6. Mechanical analysis
The mechanical properties of the hydrogels were tested using a static mechanical tester (Stable Microsystems, TA-HD plus, UK). The hydrogels were prepared in cylindrical molds. The height and diameter of the hydrogels was 20 and 15 mm, respectively. This resulted in the L/D ratio of 1.33. The hydrogels were subjected to cyclic compression, cyclic stress relaxation (SR), and cyclic creep studies to understand the viscoelastic properties of the hydrogels.[Citation16]
2.7. Impedance analysis
The electrical properties of the hydrogels were tested using an in-house built impedance analyzer in the frequency range of 200 Hz–20 kHz. The set-up was used to determine the V–I characteristic by altering the amplitude of the sinusoidal voltage signals. The frequency of the sinusoidal signal was kept constant at 10 kHz.
2.8. Biological characterization
The mucoadhesive property of the hydrogels was determined using static mechanical tester (Stable Microsystems, TA-HD plus, UK). Goat intestine was used as the representative mucosal layer for the study. The goat intestine was collected in cold saline from the local slaughter house. The intestines were longitudinally cut open and were further cut into pieces of 1 cm × 1 cm. The intestinal pieces were attached onto the base of the mechanical tester. Subsequently, the hydrogels (5 mm × 5 mm) were attached on the 30-mm flat probe using double sided acrylate tape. Thereafter, the flat probe was lowered at a speed of 0.5 mm/s, and a force of 20 g was applied on mucosal surface for 10 s to promote adhesion between the hydrogels and the mucosal layer. The probe was then retracted back at the same speed. The force required to separate the hydrogel from the intestinal mucosal surface was noted as mucoadhesive force. The work of mucoadhesion was calculated from the area under the curve of the force–time profile.
The biocompatibility of the hydrogels was estimated by hemocompatibility and cell viability test. The hemocompatibility test dealt with the incubation of the hydrogels in diluted goat blood. The percentage hemolysis of the goat blood was calculated from the absorbance of the supernatant fluid obtained after centrifuging the goat blood containing the hydrogel pieces.
The cytocompatibility of the hydrogels were determined using MG63 cells. The cells were seeded in 96-well plates. 1 × 104 cells were added in each well. Twenty microliter of leachants (of hydrogels) was added in each well to understand the toxic effect of the leachants. The cell viability was determined using MTT assay.
2.9. Swelling studies
The swelling profiles of the hydrogel was determined at pH 1.2 (0.1 N HCl) and pH 7.4 (phosphate buffer). The weights of the hydrogels, immersed in the swelling media, were determined after an interval of 15 min for the first 1 h and 30 min for the next 9 h. The study was conducted at room temperature. The swelling index was calculated as per the following equation:(1)
where WT = Weight of sample at time T, and W0 = Dry weight of the sample before the start of the study.
2.10. Drug release studies
The drug release studies were carried out using accurately weighed hydrogel samples (~350 mg). The hydrogels were put in dialysis tube containing 1 ml of dissolution media. Both ends of the dialysis membrane were sealed using dialysis tube clips. The setup was lowered in a beaker containing 50 ml of dissolution media, kept under stirring at 100 rpm. The temperature of the dissolution media was maintained at 37 °C. At regular intervals of time, the dissolution media was replaced with fresh dissolution media for 10 h. The replaced media was analyzed for the drug content using UV–visible spectrophotometer (Systronics, Double beam spectrophotometer (2203), India). The study was conducted using 0.1 N HCl (pH 1.2) and phosphate buffer (pH 7.4).
The qualitative drug release study was conducted by performing antimicrobial test using disc diffusion method. E. coli was used as the test microorganism. Hydrogel samples of 9 mm diameter were used for the analysis. The antimicrobial activity was correlated with the zone of inhibition of the microbial growth.
3. Result and discussion
3.1. Preparation of hydrogels
Phase-separated hydrogels are a special class of mix polymer systems. In these hydrogels, the polymers separate out (concentrate) as individual polymeric phases. The phase separation may happen in different ways due to inter- and intra- polymeric interactions. Based on the interactions, the mix biopolymer system may form three types of molecular architectures, namely, segregative phase separation, associative phase separation, and bicontinuous phase separation.[Citation9,17] Segregative phase separation happens when the two polymers have negative associative interactions. The affinity of the polymer toward the solvent may also alter the molecular dynamics of the segregative phase-separation process. This results in the formation of two phases which are enriched with either of the polymers. The associative phase separation has been reported to occur when the interactions among the two polymers are very strong. This result in the separation of the polymer–polymer composite (dispersed phase) and the solvent forms the continuous phase. The bicontinuous phase-separated hydrogels are formed when both the polymer phases appear as continuum phase. Quite often, many scientists have regarded this class of phase-separated hydrogels as a specific category of segregative phase separation.[Citation18] Gelatin/polysaccharides-based phase-separated systems have been reported to form hydrogels by segregative phase-separation mechanism. These hydrogels are usually chemically crosslinked to improve the physical stability. This is done because the previous reports suggest that water-in-water type of emulsions have stability issues, similar to the one confronted by the oil–water emulsions.[Citation19]
In this study, it was observed that an increase in the proportion of TG was associated with the increase in the whiteness of the formulation (Figure ). It has been previously reported that the emulsions appears as white in color due to the diffraction of the light from the interface of the internal and the continuum phases. This gives the indication that there was a probability of formation of water-in-water emulsions. Similar observation was also made in the gelatin-CMT hydrogels. The whiteness of the CMT hydrogels was lower than the TG hydrogels. This observation may be explained by the fact that the carboxymethylation of TG resulted in the increased hydrophilicity of the TG backbone. The increase in the hydrophilicity might have improved the interaction among gelatin and CMT. Hence, it may be expected that the degree of phase separation will be lower as compared to the TG hydrogels. Hydrogels were smooth to touch and had a soothing effect.
Figure 2. Pictographs of the hydrogels. (a) T1, (b) T2, (c) T3, (d) C1, (e) C2, and (f) C3.

3.2. Microscopy
The gelatin polysaccharide mixture (50 °C) was converted into thin smears over a glass slide. The smear was observed under the bright-field microscope (Figure ). The micrographs of the formulation show homogeneous distribution of the globular microstructure of polysaccharides within the gelatin continuum phase. The size of the globular microstructures was found to increase as the concentration of the polysaccharides was increased in the formulations. This may be due to the higher intra-polysaccharide interactions during the gelation process. The globular size of the dispersed phase was found to be higher in TG hydrogels. Similar results were expected from the visual observation of the hydrogel matrices. The large globular size of the dispersed phase in the TG hydrogels may be accounted to the higher intra-polysaccharide interactions. Carboxymethylation of tamarind gum resulted in the formation of anionic polyelectrolyte, which in turn, resulted in the increased hydration of the polysaccharides. This resulted in the decrease in the intra-polysaccharide interactions with a subsequent increase in the inter-polymer interactions. The decrease in the intra-polysaccharide interactions may be explained by the ionization of the carboxylic groups of CMT. The ionization of the carboxylic groups resulted in the stearic hindrance, which in turn, hindered the process of self-aggregation of CMT.[Citation20]
Figure 3. Light micrographs of the hydrogels. (a) T1, (b) T2, (c) T3, (d) C1, (e) C2, and (f) C3.

3.3. Infrared spectroscopy
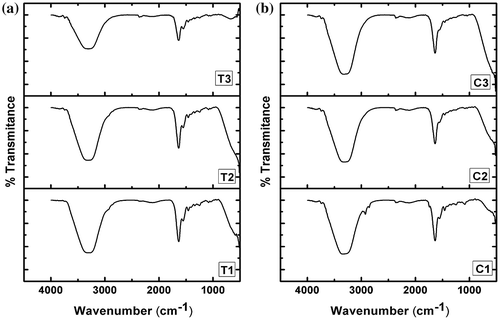
The FTIR spectra of hydrogels were acquired in the ATR mode (Figure ). The spectra of all the hydrogels were found to be similar with that of gelatin alone hydrogel as reported in the literature (Figure S1).[Citation21] Gelatin is a protein molecule. The gelatin hydrogel showed a broad peak at ~3320 cm−1. This peak may be associated with the O–H and N–H stretching vibrations. The peak at ~1650 cm−1 can be explained by C–O and C–N stretching of the amide bands. The peak at ~1550 cm−1 may be associated with amide-II bonds, whereas the peak at ~1250 cm−1 may be attributed to amide III bonds. It has been reported by various groups that the peak in the region of 1600–1700 cm−1 is an important peak for the analysis of the secondary protein structures. Addition of TG to the gelatin hydrogels did not shift the peak position of the gelain in the hydrogels. This suggested that there were no significant changes in the secondary structure of the gelatin. CMT in lower proportions did not alter the peak position at ~1629 cm−1 but at higher concentration of CMT, there was a shift in the amide-I peak toward higher wave number. Such shift in the amide-I peak has been previously explained by the interaction of the COO− groups (present in polysaccharides) with the amide-I group of the gelatin.[Citation21] Additionally, the extent of hydrogen bonding among the polysaccharide containing hydrogels was estimated by determining the area under the curve of the peak present in the region of 3700–2900 cm−1. The area under the curve of the TG hydrogels were found to be in the order of T1 ≫ T2 ≫ T3. On the other hand, the area under the curve for the CMT containing hydrogels was found to be higher for the formulations containing higher amount of CMT. In general, the area under the curve of the CMT hydrogels was higher than the TG hydrogels. The above result may be explained by the relatively hydrophobic nature of TG. An increase in the TG concentration within the formulation resulted in the predominant hydrophobic interaction among the TG molecules. This, in turn, affected the inter-molecular interactions among the TG and the gelatin molecules. On the other hand, CMT molecules interacted with the gelatin molecules due to the availability of the free carboxylic groups.
Figure 4. FTIR spectra of the hydrogels. (a) TG hydrogels and (b) CMT hydrogels.
3.4. Thermal analysis
T1, T3, C1, and C3 were taken as the representative hydrogels for the thermal analysis. The thermal profiles and the thermal parameters of the hydrogels have been given in Figure and Table , respectively. A broad peak was observed in the initial part of the thermal profiles. This peak may be explained by the evaporation of the water molecules from the hydrogels. The peak position was at ~100 °C for T1, whereas it was at ~78 °C for T3. The lowering of the endothermic peak temperature suggested that there might be a reduction in the hydrogel–water interactions. This can be explained by the presence of the hydrophobic TG in higher concentration in T3. The enthalpy and entropy of the TG hydrogels were found to be higher when the concentration of the TG was more. This suggested that the energy required to evaporate the water molecules present in the hydrogels was higher when the concentration of polysaccharides was increased.
Figure 5. Thermal analysis of the hydrogels. (a) T1, (b) T3, (c) C1 and (d) C3.
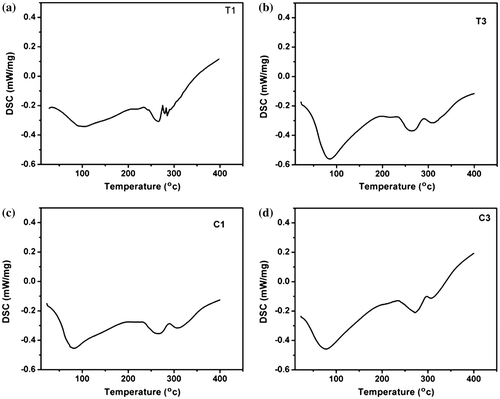
Table 2. Changes in enthalpy (ΔH) and entropy (ΔS) values of the hydrogels.
An increase in the endothermic peak temperature was observed when the concentration of the CMT was increased in CMT hydrogels. This can be explained by the relatively hydrophilic nature of CMT (as compared to TG). An increased hydrophilicity increased the interaction of the hydrogel matrices and the water molecules. The enthalpy and the entropy associated with the evaporation of the water molecules was found to be higher in hydrogels containing higher proportions of CMT. Similar changes in enthalpy and entropy were also observed in TG hydrogels. In general, the enthalpy and entropy of the CMT hydrogels were found to be higher as compared to the TG hydrogels of similar composition. This observation can also be explained by the increased hydrophilicity of CMT hydrogels due to the presence of hydrophilic carboxymethylated group. Apart from the broad endothermic peak, associated with the evaporation of the water molecules, two more broad peaks were observed at ~270 and ~315 °C. These additional peaks may be due to the degradation of the gelatin polymeric structure at higher temperatures. Similar endothermic peaks were also observed in pure gelatin matrices.
3.5. Mechanical analysis
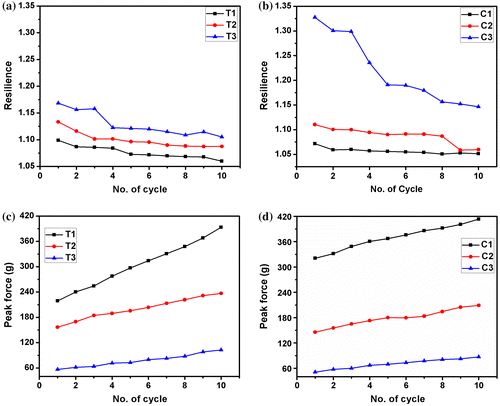
Cyclic compression test was conducted to understand the variation in the mechanical properties of the hydrogels when repeated stress was applied. In general, the peak force of the hydrogels was found to be lower in the hydrogels with higher proportion of polysaccharides. This suggested that the incorporation of the higher proportion of polysaccharides resulted in the formation of the softer hydrogels. The resilience of the hydrogels was also estimated from the cyclic compression test. Resilience of a particular compressive cycle is defined as the ratio of the area under the curve during the compression to area the under the curve during decompression.[Citation22] The resilience is a measure of the ability of the hydrogels to undergo recovery after compression. In general, the resilience was found to be higher in the hydrogels containing higher proportions of polysaccharides. After cyclic compression for 10 cycles, a non-linear reduction in the resilience was observed. The percentage decrease of resilience after 10 cycles of compression was higher in the hydrogels containing higher proportions of polysaccharides (Figure (a) and (b)). In general, a higher resilience is observed in softer material. Our observation suggests that the hydrogels with higher proportion of polysaccharides were softer in nature. Subjecting the hydrogels to repetitive compression resulted in the decrease in the softness. This can be explained by the work hardening phenomenon. Even though the peak force (first cycle) of the TG hydrogels was lower than the CMT hydrogels at lower concentration, the peak forces were found to be nearly equal when the polysaccharide content was increased. After 10 cycles of compression, an increase in the compressive strength of all the hydrogels was observed (Figure (c) and (d)). The percentage increase in the compressive strength was higher in TG hydrogels. An increase in the compressive strength upon repeated application of the stress indicated that there was an increase in the elastic component with a corresponding decrease in the viscous component. This kind of phenomenon is often regarded as work hardening.
Figure 6. Cyclic compression results of the hydrogels. (a) Resilience of TG hydrogels, (b) resilience of CMT hydrogels, (c) peak forces of TG hydrogels and (d) peak forces of CMT hydrogels.
The hydrogels were subjected to SR studies (Table , Figure ). The peak force during (F0) during the application of the stress was found to be higher in the TG hydrogels as compared to the CMT hydrogels at lowest concentration of the polysaccharide (T1 and C1). This can be accounted to the higher filler effect exerted by the phase-separated TG. An increase in the polysaccharide showed an inverse trend. This can be explained by the incorporation of the defects in the gelatin hydrogels. In general, an increase in the polysaccharide content resulted in the decrease in the F0 values. This indicated a decrease in the firmness of the hydrogels with the increase in the polysaccharide content and a corresponding decrease in the gelatin content. The percentage relaxation (% SR) was higher in the hydrogels with higher proportions of polysaccharide, suggesting an increase in the viscoelastic fluid component. % SR of the CMT hydrogels was higher. This can be correlated with the filler effect, which in turn, increased the rigidity of the TG hydrogels as compared to the CMT hydrogels.
Table 3. Stress relaxation parameters of the hydrogels.
Figure 7. Stress relaxation results. (a) Stress relaxation profiles, (b) Peleg’s analysis, (c) Wiechert model, fitting of the normalized profiles: (d) T1, (e) T2, (f) T3, (g) C1, (h) C2, and (i) C3.
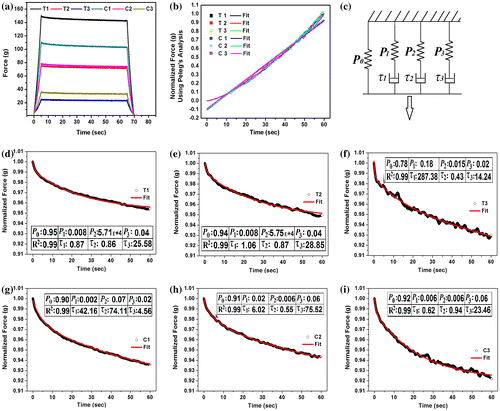
The normalized relaxation data were fitted to the Peleg’s empirical equation (Figure , Table ). The Peleg’s equation is given by:(2)
The k1 and k2 values represent the initial rate and the extent of relaxation, respectively. These parameters were calculated. The results suggested that the initial rate of relaxation in TG hydrogels was lower than the CMT hydrogels. The extent of relaxation was nearly equal in all the hydrogels.
The relaxation data were further fitted to the Weichert model of viscoelasticity (Figure (b)). The viscoelastic mathematical model and the calculated viscosity of the dashpots of the model have been tabulated in Table . The time-independent elastic modulus (P0) was dependent on the composition of the hydrogels. There was a decrease in the P0 values with the increase in the polysaccharide content in TG hydrogels. On the contrary, a reverse trend was observed in the CMT hydrogels. Hence, it can be expected that the viscous component of the TG and the CMT hydrogels will increase with the increase in the TG content and decrease in the CMT content, respectively. The validation of the results was done by calculating the viscosities of the dashpots of the Weichert model. The viscosities of the dashpots were higher in hydrogels with higher proportion of TG and lower proportion of CMT. The results were found to be as per the expectations.
Table 4. Stress relaxation model fitting using Wiechert model.
The hydrogels were further subjected to cyclic SR. The SR was found to be higher in the first cycle. As the hydrogels were subjected to repeated SR studies, a decrease in the SR was observed (Figure (a) and (b)). This indicated that during the cyclic SR studies, there was an increase in the viscoelastic solid property of the material with the subsequent decrease in the viscoelastic fluid nature. These results are concurrent with the cyclic compression studies which also suggested an increase in the viscoelastic solid component of the hydrogels as they were subjected to cyclic compression.
Figure 8. Cyclic stress relaxation results. (a) % SR of TG hydrogels, (b) % SR of CMT hydrogels, (c) D20 values of TG hydrogels, and (d) D20 values of CMT hydrogels.
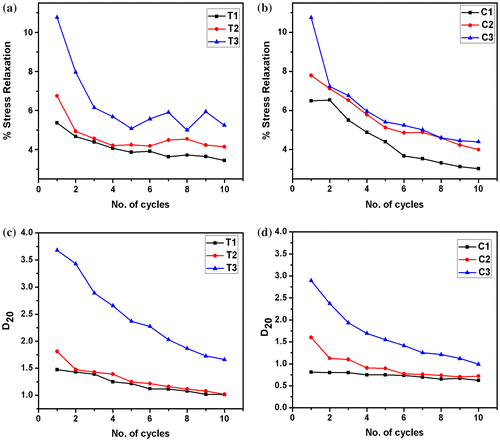
The firmness of the hydrogels was predicted by calculating the D20 values. D20 is regarded as the distance travelled by the probe to attain a force of 20 g. In general, the D20 values are inversely proportional to the firmness of the hydrogels. This is due to the fact that the firm hydrogels will resist the movement of the probe as compared to the softer hydrogels. An increase in the D20 values was observed with an increase in the polysaccharide content (Figure (c) and (d)). Among the two types of the hydrogels, CMT hydrogels showed a lower D20 values as compared to TG hydrogels. This suggested that the CMT hydrogels were relatively more firm as compared to the TG hydrogels. This result may be correlated to the extent of the H– bonding predicted from the area under the curve of the FTIR peak in the region 3700–2900 cm−1. The FTIR results showed that the AUC were comparatively higher in CMT hydrogels, suggesting a higher degree of hydrogen bonding in CMT hydrogels.
Cyclic creep and recovery studies were carried out and the changes in the viscosity (η), instantaneous creep compliance (Jo), and creep recovery were analyzed (Figure ). The hydrogels showed an increase in the viscosity after repetitive creep studies. In general, an increase in the polysaccharide content resulted in the increase in the initial viscosity of the hydrogels. The only exception being T2. The % increase in the viscosity was higher in TG hydrogels as compared to the CMT hydrogels. An increase in the concentration of TG resulted in the increase in the % increase in the viscosity. On the contrary, % increase in the viscosity of the CMT hydrogels was higher when the polysaccharide content was low. All the hydrogels showed a decrease in the creep compliance upon repetitive creep studies. A decrease in the creep compliance was observed with the increase in the proportion of CMT, whereas the creep compliance of TG hydrogels showed an initial decrease followed by an increase in the creep compliance. At lower concentration of polysaccharides, the creep compliance was higher in TG hydrogels. But when the concentration of the polysaccharides was highest (T3 and C3), the creep compliance was found to be nearly similar. As a matter of fact, creep compliance is inversely related to the stiffness of the hydrogels. This suggested that the stiffness of the CMT hydrogels was higher as compared to the TG hydrogels. Similar results were also observed from the compression and the SR studies. The % decrease in the creep compliance decreased with a corresponding increase in the polysaccharide content. TG hydrogels showed higher creep compliance as compared to the CMT hydrogels. On the other hand, a reverse trend was observed at higher proportions of the polysaccharide (T3 and C3).
Figure 9. Cyclic creep results: (a) T1, (b) T2, (c) T3, (d) C1, (e) C2, and (f) C3 and Recovery results: (g) TG hydrogels and (h) CMT hydrogels.
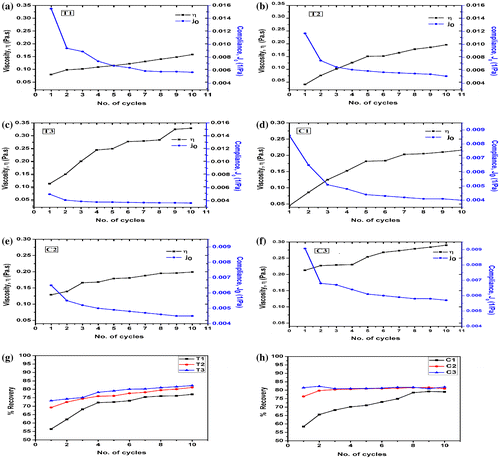
The creep recovery was calculated and analyzed. The initial recovery of both types of the hydrogels was nearly similar at lower concentration of the polysaccharides (T1 and C1). The % recovery was found to be relatively higher in CMT hydrogels with the increase in the polysaccharide content. An increase in the creep recovery profile was observed in both types of hydrogels. The % increase in the recovery was higher in TG hydrogels. An increase in the concentration of the polysaccharides resulted in the decrease in the % creep recovery after 10 cycles of creep studies.
3.6. Impedance analysis
The impedance profiles of both the types of hydrogels were found to be similar. An increase in TG content resulted in the increase in the impedance of the hydrogels (Figure (a)). This can be explained by the hydrophobic nature of the TG molecules, which promoted the formation of predominant biphasic systems. The TG molecules in the hydrogel system behaved as dielectric material. As the proportion of TG was increased, there was a corresponding increase in the size of the dispersed phase molecules. This, in turn, resulted in the increase in the dielectric behavior of the dispersed phase. In general, higher the proportion of dielectric material, higher is the impedance. An increase in the CMT concentration resulted in the decrease in the impedance of the CMT hydrogels (Figure (b)). This observation may be explained by the polyelectrolyte nature of CMT. An increase in the polysaccharide content resulted in the increase in the CMT (polyelectrolyte) content in the hydrogels, which in turn, promoted the flow of current through the hydrogel matrices. The impedance profile of the hydrogels showed higher impedance in lower frequencies. The impedance exponentially degraded to a base level at higher frequencies. This kind of impedance profile is usually associated with the capacitive nature of the materials. The higher impedance at lower frequency may be explained by the electrode polarization effect at lower frequencies. The polarization effect is minimized when the frequency of the injecting current is increased. This is due to the quick reversal of the electrode polarity at higher frequencies. This phenomenon prevents the polarization effect at the electrode–sample interface.[Citation23]
Figure 10. Impedance profiles: (a) TG hydrogels and (b) CMT hydrogels and V–I characteristics: (c) TG hydrogels and (d) CMT hydrogels.
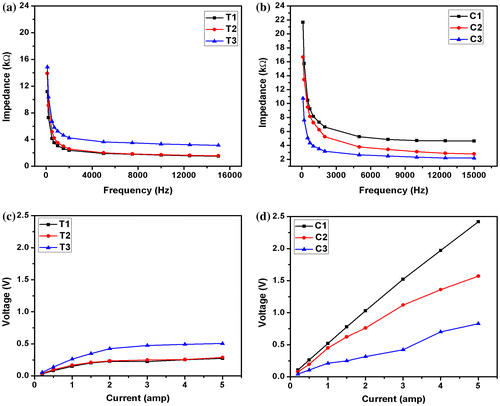
The V–I characteristics of the hydrogels were measured at 10 kHz. This was done to eliminate the electrode polarization effect of the electrodes. The V–I profile of the TG hydrogels showed an initial linear increase in the voltage when the current was increased (Figure (c)). Subsequently, the V–I profiles became saturated as the current was increased further. This may be explained by the internal alteration of the microstructure of the hydrogels when the current was increased beyond a critical value. On the other hand, the V–I profiles of the CMT hydrogels followed a linear profile (Figure (d)). This indicated that the CMT hydrogels behaved as pure resistive formulation under the experimental conditions. The slope of the V–I curve gives an indication about the impedance of the material. Similar to the impedance profile, the V–I profile also suggested that an increase in TG resulted in the increased impedance of the TG hydrogels. On the contrary, the impedance of the CMT hydrogels was found to be lower in hydrogels containing higher proportions of CMT.
3.7. Biological characterization
Both gelatin and polysaccharides have been reported to be excellent materials for designing mucoadhesive systems.[Citation24] Mucoadhesion depends on the type of functionalization and the method of preparation/extraction of the above biopolymers. In this study, the mucoadhesive property was determined using a static mechanical tester. An increase in the TG content showed an increase in the mucoadhesion of the TG hydrogels (Figure (a)). This can be explained by the composition of the hydrogels. In TG hydrogels, incorporation of TG (a non-ionic polysaccharide) resulted in the increase in the number of the hydroxyl groups which have the capability to positively interact with the mucin. On the other hand, an increase in the CMT content resulted in the decrease in the mucoadhesion (Figure (a)). This is due to the fact that the mucosal layer mainly consists of mucin (negatively charged polysaccharide).[Citation25] The negative charge of the mucin resulted in the repulsion of the carboxylic groups present in the CMT hydrogels. This resulted in a negative interaction between the mucosal layer and the CMT hydrogels. Among the hydrogels of similar composition, T1 showed lower mucoadhesive properties than C1, whereas T2 and C2 showed similar mucoadhesive properties. This observation may be explained by the interaction of the polysaccharides with the gelatin matrix.
Figure 11. Biological characterization of the hydrogels. (a) Area under the curve of mucoadhesive profiles, (b) % hemolysis of goat blood, (c) cell viability study and (d) antimicrobial study.
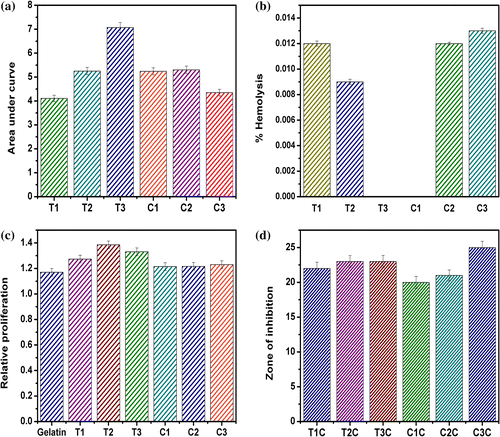
The biocompatibility of the hydrogels was determined by hemocompatibility and cytocompatibility tests. The hemocompatibility study is based on the determination of the lysis of RBC in the presence of the hydrogels. The lysis of RBC results in the release of the hemoglobin in the external fluid. The hemoglobin gets solubilized in the external fluid and gives a yellowish color. The suspended unwanted components of the blood are pelletized by centrifugation. The yellowish color of the supernatant fluid is measured using a UV–visible spectrophotometer and is compared with the positive and negative controls to determine the percentage hemolysis of the RBCs in the presence of the hydrogels. All the hydrogel formulations were found to be highly hemocompatible as the percentage hemolysis in the presence of hydrogels was ≪5% (Figure (b)).
The cytocompatibility of the hydrogels was checked using MG63 cell line. Extract of the hydrogels were prepared and were used for the analysis. The experiment was done to test whether the leachants of the hydrogels have any adverse effect on the proliferation of the cells. The relative proliferation of all the hydrogels was >1, which indicated the cytocompatible nature of the hydrogels (Figure (c)). It may also be concluded that the prepared hydrogels supported the growth of MG63 cells to a certain extent as compared to the control. In general, TG hydrogels showed better cell growth as compared to CMT hydrogels.
The antimicrobial activity of the drug-loaded hydrogels was estimated against E. coli. The blank hydrogels did not show any zone of inhibition but samples loaded with the antimicrobial drug showed a marked zone of inhibitions (Figure (d)). This suggested that the blank hydrogels did not contain any antimicrobial activity, whereas a marked antimicrobial activity of the drug-loaded hydrogels was evident. The result suggested that the drug was released from the hydrogels matrices in active form.
3.8. Swelling studies
The swelling studies were conducted at pH 1.2 and 7.4 (Figure (a)–(d)). At both the pHs, the swelling of the TG hydrogels was found to be higher in the formulations containing higher proportion of TG. The swelling index of the TG hydrogels was higher at pH 1.2. T1 and T2 showed similar swelling at pH 1.2. On the contrary, the swelling was found to be markedly different at pH 7.4. Higher swelling at pH 1.2 may be explained by the ionization of the amino groups of gelatin molecules. This resulted in the electrostatic repulsion of the gelatin polymer chains. This, in turn, resulted in the exposure of the polysaccharide phase in the matrices. The inflow of the water through the more porous gelatin polymer network resulted in the increased wetting of the polysaccharide phase. Due to this reason, the swelling was higher at pH 1.2. On the contrary, at near neutral pH (pH 7.4), the carboxylic and amino groups existed as zwitterions. This resulted in the existence of the gelatin network as a neutral polymeric architecture. Hence, the swelling of the matrices was dependent mainly on the absorption of the water molecules by the polysaccharide phase. The above phenomenon resulted in the marked differences in the absorption of the water molecules with the variation in the polysaccharide content.
Figure 12. Swelling behavior of the hydrogels: (a) TG hydrogels (pH 1.2), (b) CMT hydrogels (pH 1.2), (c) TG hydrogels (pH 7.4), (d) CMT hydrogels (pH 7.4), and drug release studies: (e) TG hydrogels (pH 1.2), (f) CMT hydrogels (pH 1.2), (g) TG hydrogels (pH 7.4), and (h) CMT hydrogels (pH 7.4).
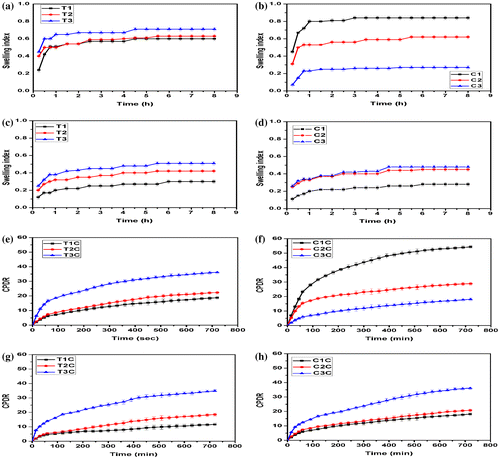
The swelling profile of the CMT hydrogels suggested a pH-dependent swelling behavior. At pH 1.2, the hydrogels containing lower amount of CMT showed higher swelling and vice versa. The swelling of these hydrogels may be explained by the combined electrostatic phenomena occurring at both the polymeric phases. The amino groups of the gelatin were ionized at pH 1.2, whereas the shielding effect was more predominant in the polysaccharide phase due to the presence of the free carboxylic groups. At lower proportions of CMT, there was a decrease in the size of polysaccharide phase, which created space for the absorption of additional amount of swelling media. On the contrary, when the polysaccharide content was increased in the hydrogels, the stearic hindrance exerted by the polysaccharide phase to the diffusion of the dissolution media was more predominant. This phenomenon can explain the decrease in the swelling at pH 1.2 of the hydrogels as the polysaccharide content was increased. A reverse swelling behavior trend was observed at pH 7.4. In other words, the swelling was found to be higher in the hydrogels, containing higher proportions of polysaccharide. The swelling behavior at this pH of the CMT hydrogels was found to be similar as that of TG hydrogels. The result suggested that the incorporation of the anionic polyelectrolytes showed a mark difference in the swelling of the CMT hydrogels at lower pH. The difference was found to be negligible at near neutral pH.
3.9. Drug release study
The swelling studies of the hydrogels indicated a possibility of pH-dependent drug release from the phase-separated hydrogels. In general, the release of the drug from the hydrogels is dependent on the swelling property of the hydrogels. Hence, to understand the pH sensitive drug release behavior from the hydrogels, the drug release studies were conducted at pH 1.2 (gastric pH) and pH 7.4 (average intestinal pH) (Figure (e)–(h)). The release of the drug from the TG hydrogels was higher from the hydrogels with higher TG content, i.e. cumulative percentage of drug release (CPDR) was in the order of T1 < T2 < T3. Similar CPDR values were also observed at pH 7.4. The CPDR of the CMT hydrogels at pH 1.2 was in order of C1 > C2 > C3, whereas the release at pH 7.4 was found to be in the order of C3 > C2 > C1. The observed result from the phase-separated hydrogels can be correlated and well explained by the swelling studies. In general, the release of the drugs from the hydrogel matrices depends on the rate of diffusion of the dissolution media into the matrices. Higher flow of dissolution media into the hydrogels results in the increased dissolution of the drug and consequently increased diffusion of the drug from the polymer matrices. A visual observation of the release studies from the hydrogels suggested that C3 may be tried as a delivery vehicle for enteric drug delivery. Usually, the enteric drug delivery vehicles show a CPDR of less than 10% in gastric pH (pH 1.2).[Citation26] The release studies suggested that the release of C3 hydrogels was below 10% up to 180 min, which is much longer than the reported literature.
A critical analysis of the drug release kinetics was carried out. The result suggested that the release of the drug from the hydrogels, under the both pH conditions, followed Higuchian release kinetics. This suggested that the developed hydrogels were behaving as planner matrices and the drug-loaded hydrogels may be regarded as matrix-type delivery vehicles. Subsequently, the diffusion mechanism of the drug from the hydrogels was estimated by calculating the diffusion value (n) using Korsmeyer–Peppas diffusion model. The ‘n’ values for the TG hydrogels (except T1) were found to be >0.45 at pH 7.4. The ‘n’ value of T1 was 0.41 at pH 7.4. The ‘n’ value suggested that release of the drug from the hydrogels except (T1) was non-Fickian diffusion mediated, whereas the diffusion from T1 was Fickian diffusion mediated.[Citation27 The ‘n’ value was found to be >0.45 for all the CMT hydrogels except C3 at pH 1.2. The value was <0.45 in C3. This suggested that the diffusion of the drugs (except C3) followed non-Fickian diffusion kinetics, whereas the release of the drug from C3 was Fickian diffusion mediated.[Citation27]
4. Conclusion
This study delineates the development of TG- and CMT-based phase-separated hydrogels. In this study, gelatin was used as the second polymeric phase. An increase in the hydrophilicity of TG by carboxymethylation resulted in the increase in the compatibility of the polysaccharide and the gelatin phase. This resulted in the better mechanical properties of the CMT-based hydrogels. A higher proliferation of the MG63 cells was observed in the presence of the extracts of TG hydrogels. The CMT-based hydrogels also showed higher proliferation as compared to the controls. Both types of the hydrogels also showed good mucoadhesive properties. Due to the presence of the free carboxylic groups in CMT, a pH sensitive swelling and drug release behavior from the CMT hydrogels were observed.
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.10.1080/15685551.2015.1041075.
Figure_S1-1.tif
Download TIFF Image (1.1 MB)Disclosure statement
The authors certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
Additional information
Funding
References
- Sechriest VF, Miao YJ, Niyibizi C, Westerhausen-Larson A, Matthew HW, Evans CH, Fu FH, Suh JK. GAG‐augmented polysaccharide hydrogel: a novel biocompatible and biodegradable material to support chondrogenesis. J. Biomed. Mater. Res. 2000;49:534–541.10.1002/(ISSN)1097-4636
- Gref R, Rodrigues J, Couvreur P. Polysaccharides grafted with polyesters: novel amphiphilic copolymers for biomedical applications. Macromolecules. 2002;35:9861–9867.10.1021/ma021132a
- Dufresne A. Comparing the mechanical properties of high performances polymer nanocomposites from biological sources. J. Nanosci. Nanotechnol. 2006;6:322–330.
- Ciardelli G, Chiono V, Vozzi G, Pracella M, Ahluwalia A, Barbani N, Cristallini C, Giusti P. Blends of poly-(ε-caprolactone) and polysaccharides in tissue engineering applications. Biomacromolecules. 2005;6:1961–1976.10.1021/bm0500805
- Jani GK, Shah DP, Prajapati VD, Jain VC. Gums and mucilages: versatile excipients for pharmaceutical formulations. Asian J. Pharm. Sci. 2009;4:309–323.
- Leakey RR. Potential for novel food products from agroforestry trees: a review. Food Chem. 1999;66:1–14.10.1016/S0308-8146(98)00072-7
- Picout DR, Ross-Murphy SB, Errington N, Harding SE. Pressure cell assisted solubilization of xyloglucans: tamarind seed polysaccharide and detarium gum. Biomacromolecules. 2003;4:799–807.10.1021/bm0257659
- Sharma M, Mondal D, Mukesh C, Prasad K. Preparation of tamarind gum based soft ion gels having thixotropic properties. Carbohydr. Polym. 2014;102:467–471.10.1016/j.carbpol.2013.11.063
- Kulkarni RV, Mutalik S, Mangond BS, Nayak UY. Novel interpenetrated polymer network microbeads of natural polysaccharides for modified release of water soluble drug: in-vitro and in-vivo evaluation. J. Pharm. Pharmacol. 2012;64:530–540.10.1111/jphp.2012.64.issue-4
- Sano M, Miyata E, Tamano S, Hagiwara A, Ito N, Shirai T. Lack of carcinogenicity of tamarind seed polysaccharide in B6C3F1 mice. Food Chem. Toxicol. 1996;34:463–467.10.1016/0278-6915(96)87356-X
- Goyal P, Kumar V, Sharma P. Carboxymethylation of tamarind kernel powder. Carbohydr. Polym. 2007;69:251–255.10.1016/j.carbpol.2006.10.001
- Tomlin J, Read N. The relation between bacterial degradation of viscous polysaccharides and stool output in human beings. Br. J. Nutr. 1988;60:467–475.10.1079/BJN19880119
- Ross K, Pyrak-Nolte L, Campanella O. The effect of mixing conditions on the material properties of an agar gel – microstructural and macrostructural considerations. Food Hydrocolloids. 2006;20:79–87.10.1016/j.foodhyd.2005.01.007
- Doublier J-L, Garnier C, Renard D, Sanchez C. Protein–polysaccharide interactions. Curr. Opin. Colloid Interface Sci. 2000;5:202–214.
- Tolstoguzov V. Compositions and phase diagrams for aqueous systems based on proteins and polysaccharides. Int. Rev. Cytol. 1999;192:3–31.10.1016/S0074-7696(08)60520-3
- Cloyd JM, Malhotra NR, Weng L, Chen W, Mauck RL, Elliott DM. Material properties in unconfined compression of human nucleus pulposus, injectable hyaluronic acid-based hydrogels and tissue engineering scaffolds. Eur. Spine J. 2007;16:1892–1898.10.1007/s00586-007-0443-6
- Lorén N, Langton M, Hermansson A-M. Confocal laser scanning microscopy and image analysis of kinetically trapped phase-separated gelatin/maltodextrin gels. Food Hydrocolloids. 1999;13:185–198.10.1016/S0268-005X(98)00083-6
- Jara F, Pérez OE, Pilosof AM. Impact of phase separation of whey proteins/hydroxypropylmethylcellulose mixtures on gelation dynamics and gels properties. Food Hydrocolloids. 2010;24:641–651.10.1016/j.foodhyd.2010.03.005
- Oakenfull D, Pearce J, Burley R. Protein gelation. In: Damodaran S, editor. Food proteins and their applications. Vol. 80. New York: CRC Press; 1997. p. 111–142.
- Huang L, Reekmans G, Saerens D, Friedt J-M, Frederix F, Francis L, Muyldermans S, Campitelli A, Hoof CV. Prostate-specific antigen immunosensing based on mixed self-assembled monolayers, camel antibodies and colloidal gold enhanced sandwich assays. Biosens. Bioelectron. 2005;21:483–490.10.1016/j.bios.2004.11.016
- Pranoto Y, Lee CM, Park HJ. Characterizations of fish gelatin films added with gellan and κ-carrageenan. LWT-Food Sci. Technol. 2007;40:766–774.10.1016/j.lwt.2006.04.005
- De La Cochetiere M, Durand T, Lepage P, Bourreille A, Galmiche J, Dore J. Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J. Clin. Microbiol. 2005;43:5588–5592.10.1128/JCM.43.11.5588-5592.2005
- Bammou L, Mihit M, Salghi R, Bouyanzer A, Al-Deyab S, Bazzi L, Hammouti B. Inhibition effect of natural Artemisia oils towards tinplate corrosion in HCL solution: chemical characterization and electrochemical study. Int. J. Electrochem. Sci. 2011;6:1454–1467.
- Smart J, Kellaway I, Worthington H. An in‐vitro investigation of mucosa‐adhesive materials for use in controlled drug delivery. J. Pharm. Pharmacol. 1984;36:295–299.10.1111/jphp.1984.36.issue-5
- Smirnov A, Tako E, Ferket P, Uni Z. Mucin gene expression and mucin content in the chicken intestinal goblet cells are affected by in ovo feeding of carbohydrates. Poultry Sci. 2006;85:669–673.
- Paliwal R, Rai S, Vaidya B, Khatri K, Goyal AK, Mishra N, Mehta A, Vyas SP. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomed. Nanotechnol. Biol. Med. 2009;5:184–191.10.1016/j.nano.2008.08.003
- Serra L, Doménech J, Peppas NA. Drug transport mechanisms and release kinetics from molecularly designed poly(acrylic acid-g-ethylene glycol) hydrogels. Biomaterials. 2006;27:5440–5451.10.1016/j.biomaterials.2006.06.011