
Abstract
Poly (diethyl vinylphosphonate-co-2-chloroethyl methacrylate) (P(DEVP-co-CEMA)) copolymers are synthesized by free radical copolymerization initiated by benzoyl peroxide. The reactivity ratios for the free radical copolymerization of CEMA (M1) and DEVP (M2) are r1 = 19.45 and r2 = 0.11, respectively. The desired amphiphilic copolymers with relatively low polydispersity index (PDI) and different DEVP contents are isolated and purified from the reaction mixtures through precipitation fractionation. The structures and molecular characteristics of the obtained products are characterized by 1H NMR, FTIR, and SEC analysis. All of these fractionated copolymers have a unimodal distribution and moderate PDI. The thermal properties of the P(DEVP-co-CEMA) copolymers are investigated by TGA and differential scanning calorimetry (DSC) measurements. Single glass transition temperature (Tg) appears in the DSC profiles differing from the existence of two Tgs in the blend of poly (diethyl vinylphosphonate) and poly (2-chloroethyl methacrylate), which indicates the random structure of the copolymers. The MALDI–ToF mass analysis further reveals that DEVP and CEMA units are randomly distributed in the copolymer chains. The self-assembly behavior of the P(DEVP-co-CEMA) copolymers in aqueous solution is preliminarily investigated.
Introduction
Phosphorus-containing polymers are particularly attractive for their extensive application in fuel cells,[Citation1–6] flame retardant [Citation7–10], and the biomedical field.[Citation11,12] Phosphorus-containing polymers include genetic materials DNA and RNA,[Citation13] and synthetic polymers functionalized at the main chain (e.g. polyphosphazene [Citation14,15] polyphosphoesters [Citation16]) and those functionalized at the side chain. Vinylphosphonate is one of the simplest vinyl monomers with the phosphonate groups at the side chain. Since the late 1940s, different routes for vinylphosphonate synthesis have been reported.[Citation17–21] Researches on the polymerization of vinylphosphonates are following shortly after.[Citation21,22] So far, quite a number of reports have been published investigating the polymerization of vinylphosphonates with the methods of radical,[Citation17,21–24] anionic,[Citation25–27] coordinative-anionic,[Citation28] and living rare earth metal-mediated group transfer polymerization.[Citation29–38] Among these techniques, coordinative-anionic polymerization and living rare earth metal-mediated group transfer polymerization are efficient to generate poly(vinylphosphonates) with higher molecular weight and higher conversions. Except for poly(vinylphosphonic acid) (which can be obtained in high yield and moderate molecular weight by free radical polymerization),[Citation39,40] relatively few investigations into the homo and copolymerization of vinylphosphonate monomers via radical polymerization have been reported. The few reports show that vinylphosphonate monomers fail to homopolymerize to high molecular weight products by the method of radical polymerization (especially for diethyl vinylphosphonate (DEVP)), which usually lead to low yields and provide polymers of low molecular weight.[Citation17,21–24] In the case of radical copolymerization, the final results were either unsuccessful or afforded insufficient characterization.[Citation24,29,41,42] Arcus reported the copolymerization of DEVP with styrene initiated by tert-butyl hydroperoxide and determined the reactivity ratios of this monomer pair. For relatively low DEVP contents in the copolymers, DEVP is reluctant to enter the growing polymer chain as compared to styrene.[Citation42,43] Except for styrene, radical copolymerizations of DEVP with ethylene glycol dimethacrylate,[Citation41] methyl methacrylate ,[Citation44] acrylonitrile,[Citation44] and trimethoxyvinylsilane [Citation24] are achieved, but afford inadequate characterization. Therefore, radical copolymerizations of DEVP with other monomers to get relatively high DEVP-containing copolymers with clearly characterized structure are still a significant and challenging issue.
The monomer 2-chloroethyl methacrylate (CEMA) is of special interest as halogen-functionalized comonomer for easy modification and post cross-linking.[Citation45–47] With initiating haloalkyl groups, graft copolymers can be prepared via atom transfer radical polymerization (ATRP).[Citation48] The chlorine atoms can be readily substituted by nucleophiles, for instance, azide groups, and then followed by click coupling of alkyne end-functionalized polymers to gain new properties.[Citation49,50] Therefore, CEMA is a potential comonomer for DEVP to broaden the applications of phosphorus-containing polymers. To date, to the best of our knowledge, there is no report on the copolymerization of DEVP with CEMA. What is more, due to the existence of both hydrophobic units (CEMA) and hydrophilic units (DEVP), the copolymers are able to self-assemble into nanoparticles in aqueous solution, which are promising in the fields such as microreactors and encapsulation of guest molecules. The modified copolymers show great potential in biomedical fields for polyphosphonates and proved to be high biocompatibility and adjustable degradability.[Citation11,12] Meanwhile, the chemical incorporation of a phosphorus-containing flame retardant into a methacrylate polymer via copolymerization may eliminate the leaching and heterogeneous problems associated with the additive approach.[Citation9,51]
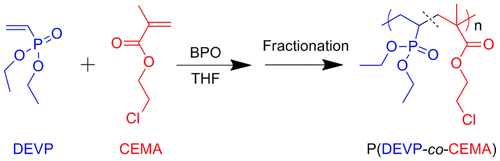
Herein, we synthesize a series of amphiphilic random copolymers, namely poly (diethyl vinylphosphonate-co-2-chloroethyl methacrylate) (P(DEVP-co-CEMA)), through free radical copolymerization using benzoyl peroxide (BPO) as initiator. The synthesis route of these random copolymers is presented in Scheme . Copolymers with relatively narrow molecular weight distribution are isolated through precipitation fractionation for the detailed investigation. The structures of the copolymers are confirmed by 1H NMR, 31P NMR, Fourier transform infrared (FTIR), and MALDI–ToF mass analysis. The thermal properties of P(DEVP-co-CEMA) copolymers are determined by differential scanning calorimetry (DSC) and thermogravimetric analyses (TGA) measurements. Preliminary investigations on self-assembly behaviors of the copolymers in water are also performed, and the nanoparticles formed in water are observed by TEM.
Experimental
Materials
Triethyl phosphite (98.0%, Aladdin Reagent, China), ethylene bromide (99.0%, Aladdin Reagent, China), triethylamine (Sinopharm Chemical Reagent, China), benzene (Sinopharm Chemical Reagent, China), methacryloyl chloride (J&K Chemical Reagent, China), and 2-chloroethanol (Shanghai Nanxiang Reagent, China) were used as received. BPO (Shanghai Lingfeng Chemical Reagent, China) was recrystallized from methanol twice before use. THF was refluxed over potassium/benzophenone ketyl before use. All the other chemicals were used as received unless otherwise specified.
Synthesis of CEMA
2-chloroethanol (10 mL) and triethylamine (22 mL) were dissolved in THF (50 mL), then methacryloyl chloride (15 mL) was added slowly under argon atmosphere while cooling the solution in an ice bath. The reaction mixture was stirred for 48 h at room temperature and then filtered. The filtrate was washed thoroughly with deionized water and dried with anhydrous sodium sulfate. The pure product CEMA was obtained by distillation under reduced pressure as colorless liquid, bp: 38–40 °C/0.1 mbar. 1H NMR (400 MHz, CDCl3, δ): 6.16 (s, 1H, CH2=), 5.61 (s, 1H, CH2=), 4.40 (t, 2H, –OCH2–), 3.73 (t, 2H, –CH2Cl), 1.95 ppm (s, 3H, –CH3). 13C NMR (400 MHz, CDCl3, δ): 166.9 (s, C=O), 135.8 (s, CH), 126.3 (s, CH2), 64.2 (s, CH2O), 18.2 ppm (s, CH2Cl).
Synthesis of DEVP
DEVP was prepared using the synthetic route we had reported before.[Citation28] Raw diethyl 2-bromoethylphosphonate (165.7 g) was prepared in 79% yield from triethyl phosphite (142.9 g) and ethylene bromide (327.4 g), and converted in 48% yield into DEVP by Kosolapoff’s methods.[Citation21] Then the crude product was distilled at 60–62 °C/0.1 mbar, and pure DEVP (60 g) was obtained. 1H NMR (400 MHz, CDCl3, δ): 1.34 (t, 6H, –CH3), 4.10 (m, 4H, –CH2O–), 6.19 ppm (m, 3H, –CH=CH2). 13C NMR (400 MHz, CDCl3, δ): 135.4 (s, CH2), 126.8, 125.0 (d, PCH), 61.8 (d, CH2O), 16.3 (d, CH3). 31P NMR (400 MHz, CDCl3, δ): 17.2 ppm.
Synthesis and fractionation of P(DEVP-co-CEMA) copolymer
A solution of DEVP (0.508 g), CEMA (0.233 g), and BPO (8 mg) in THF (3.5 mL) was introduced in a sealed tube and degassed by means of three freeze/pump/thaw cycles. The reaction mixture was then placed in a preheated (60 °C) oil bath; the reactant was allowed to cool to room temperature after reacted for 24 h, and the crude copolymer was precipitated in hexane. The desired amphiphilic P(DEVP-co-CEMA) copolymer was obtained by further precipitation fractionation using methanol/THF as nonsolvent-solvent system. The crude copolymer was dissolved in 5 mL THF, and then 50 mL methanol was added to the solution. Finally, the mixture became turbid, and the higher MW precipitate was separated through centrifugation. After centrifugation, the supernatant clear liquid was transferred to a round-bottom flask, and the organic solvents were removed using rotary evaporator. The residuum in flask was dissolved in THF, precipitated in hexane, and repeated three times. The final product was then filtered and dried under vacuum, to give a white powder.
Preparation of a self-assembly system
The assemblies were prepared using the typical procedure as follows: 2 mg copolymer was dissolved in 1 mL THF and followed by the addition of water dropwise to the desired water contents, then dialyzed against deionized water for three days to exclude the organic solvent. Regenerated cellulose dialysis tubes were used, and the specific molecular weight cut-off is 1000.
Measurements
1H NMR, 13C NMR, and 31P NMR spectra were recorded on Bruker Avance III 400 spectrometers using CDCl3 as solvent and tetramethylsilane as internal reference. FTIR spectra were obtained with a Bruker Vector 22 FTIR spectrometer by signal averaging a total of 32 scans. Thin films were prepared by casting polymer/THF solutions onto KBr discs. Size exclusion chromatography (SEC) was performed on a Waters-150C apparatus in THF at a flow rate of 1.0 mL min−1 at 40 °C using narrow MWD polystyrene standards. DSC analyses were performed on a TA Q20 instrument. Samples were heated from −80 to 200 °C at a rate of 10 °C min−1 under a nitrogen purge, quenched to −75 °C and subjected to a second scan. The second thermo-scanning curves were recorded. TGA were performed on a TA Q50 instrument. Samples were first holding for 10 min at 150 °C and then heated from 50 to 600 °C at a rate of 10 °C min−1 under a nitrogen purge. The hydrodynamic diameters of the nanoparticles were measured by dynamic light scattering (DLS) using a particle size analyzer (Zetasizer Nano Series, Malvern Instruments) at 25 °C. The measurements were taken at a fixed angle at 90° and a wavelength of 657 nm. The morphologies of nanoparticles in water were observed on JEOL JEM-1230 (HR) TEM instrument with the accelerating voltage of 80 kV. The samples were prepared by placing the solution on copper grids coated with carbon-film. After the solvent evaporated naturally, they were stained with one drop of phosphotungstic acid aqueous solution and dried under ambient condition. Matrix-assisted laser desorption/ionization time of fight (MALDI–ToF) mass spectra were collected on a Bruker UltraFLEX MALDI–ToF mass spectrometer. All polymer (3–5 mg mL−1 in THF) mass spectra were recorded in the reflection mode with an acceleration voltage of 20 kV. Dithranol (36 mg mL−1 in THF) was used as a matrix.
Results and discussion
Synthesis and characterization of P(DEVP-co-CEMA)
CEMA and DEVP monomers were prepared according to the procedures described in literatures [Citation18,42,50,52] and our previous work.[Citation28] The structures of the monomers are clearly confirmed by 1H NMR, 13C NMR, and 31P NMR (Figures S1–S5 in Supplementary Information). Homo and copolymerization of DEVP and CEMA were carried out in THF at 60 and 50 °C initiated by BPO, and the results are summarized in Table . CEMA showed high reactivity for radical homopolymerization, and poly (2-chloroethyl methacrylate) (PCEMA) with high yield (85.6%) and high MW (67.0 kDa) was prepared (Sample P9 in Table ), while only oligomers (0.4 kDa) were obtained in the case of DEVP homopolymerization (Sample P8 in Table ). This low reactivity of DEVP is consistent with the results reported before.[Citation17,21–24] The structures of PCEMA and PDEVP are confirmed by 1H NMR (Figures S6 and S7) measurements. However, P(DEVP-co-CEMA) copolymers were obtained successfully by copolymerization of DEVP and CEMA; the copolymer was precipitated in hexane, which is the poor solvent for both DEVP and CEMA segments. The copolymerization yields vary between 26.6% (Sample P3) and 65.8% (Sample P4), and decrease noticeably with the increasing of initial DEVP feed molar ratios, due to the low reactivity of DEVP. The reaction temperature also influences the copolymerization results obviously. Lower temperature (50 °C) leads to higher MWs, but lower yields and DEVP contents (Sample P1–3, Table ). From Table1, it can be seen that MWs decrease sharply with the increasing of the DEVP feed molar ratios, while DEVP contents in the copolymer increase correspondingly.
Table 1. Homo and copolymerization of DEVP and CEMA initiated by BPOTable Footnotea.
The 1H NMR spectrum (Figure (A)) demonstrates the presence of DEVP units in copolymer chains, which can be confirmed from the signals lined out at 4.1 and 1.3 ppm assigned to –OCH2– and –OCH2CH3 in DEVP segments. The 31P NMR spectra (Figures S8–S13) show signals in the 28–36 ppm region, these results further confirm the presence of DEVP units. The characteristic absorption bands of DEVP units (υP–O = 1027 and 1052 cm−1, υP=O = 1228 cm−1) also appear clearly in the FTIR spectrum (Figure (A)). The copolymer compositions (DEVP:CEMA) range from 0.005:1 to 0.42:1 (molar ratio, Table ), which deviates much from the feed molar ratios for the low reactivity of the DEVP. DSC analyses indicate that all of the copolymers prepared have single glass transition temperature (Tg), which ranges from 72.7 to 33.4 °C and decrease with the increasing of DEVP contents (Table ). For instance, DSC profile of Sample P1 (Figure (A)) shows that the copolymer has a single Tg around 71 °C differing from the appearance of two Tgs in the blend of PCEMA and PDEVP (Figure (C)), which indicates the formation of the copolymers.
Figure 1. 1H NMR spectra of Sample P1 (A) and Sample F1 (B).
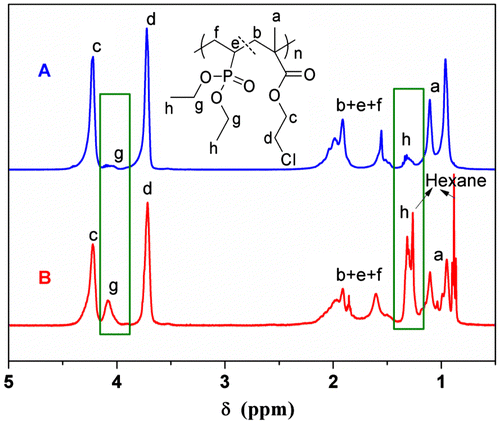
Figure 2. FTIR spectra of Sample P1 (A) and Sample F1 (B).
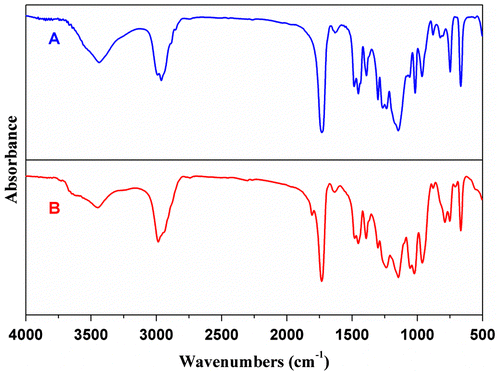
Figure 3. DSC profiles of Sample P1 (A), F1 (B), and the blend of PCEMA and PDEVP (C).
To quantify the reactivity difference between the two monomers, reactivity ratios of CEMA and DEVP were determined by the Finemann and Ross method,[Citation53] which was based on the results of 1H NMR analysis. The monomer reactivity ratios for the free radical copolymerization of CEMA (M1) and DEVP (M2) are r1 = 19.45 and r2 = 0.11 (Table S1 and Figure S14), indicating that the copolymerization should produce long blocks of CEMA and randomly distributed short blocks of DEVP.
Although the P(DEVP-co-CEMA) copolymers have been synthesized successfully, and the structures of copolymers have been confirmed by NMR, FTIR, and DSC measurements, SEC traces (Figure (A), Figures S15A–S20A) of the unfractionated copolymers precipitated in hexane show multicomponents and quite broad MW distribution. To obtain the copolymer with higher DEVP content and narrower MW distribution, methanol and THF are selected as the nonsolvent-solvent system for the precipitation fractionation. DEVP-rich copolymer fractions with comparatively narrow MW distribution are isolated and purified according to the methods described in the experimental part, and the results are listed in Table . For instance, the fractionated copolymer sample F1 has a higher DEVP:CEMA (0.060:1) and a narrower MW distribution polydispersity index (PDI = 1.30) as compared to sample P1 (DEVP:CEMA = 0.005:1, PDI = 3.81) according to the 1H NMR spectrum (Figure (B)) and SEC trace (Figure (B)).
Figure 4. SEC traces of Sample P1 (A) and Sample F1 (B).
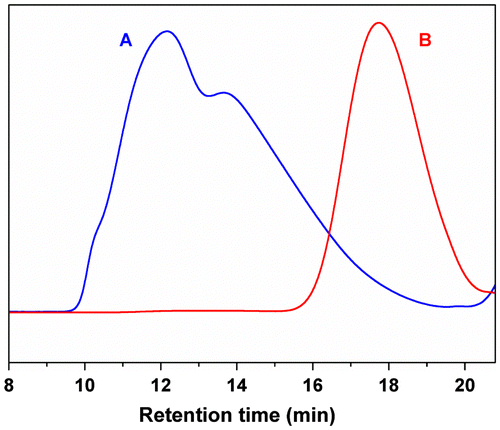
Table 2. Characterization of low MW fractions of P(DEVP-co-CEMA)Table Footnotea.
Due to the fractionation, the MWs and polydispersities of the copolymers fractionated are not much different. SEC measurements indicate that all the copolymers have a similar unimodal distribution after fractionation as shown in Figure (B), as well as other samples shown in Supplementary Information (Figures S15B–S20B). The MWs of the copolymers are ranging from 2.4 to 4.6 kDa. All the copolymers of CEMA and DEVP exhibit moderate PDIs (1.31–1.48). The copolymer compositions (DEVP:CEMA) range from 0.06:1 to 0.42:1 (molar ratio, Table ), which are also lower than the initial monomer feed ratios, but still remain positive relationship. The chemical structures of copolymer P(DEVP-co-CEMA) are confirmed by FTIR and 1H NMR analyses. The FTIR spectrum of P(DEVP-co-CEMA) copolymer (Sample F1 in Table ) is shown in Figure (B). The characteristic absorption bands of CEMA unit (υC=O = 1750 cm−1, υC–O = 1140 cm−1, υC–Cl = 745 cm−1) appear clearly in the FTIR spectrum. The presence of peaks at 1228, 1027, and 1052 cm−1 is attributed to the P = O, P–O stretching vibration of DEVP unit. The 1H NMR spectrum of the P(DEVP-co-CEMA) copolymer (Figure (B)) exhibits signals from methylene and methine protons in the backbone of both CEMA and DEVP units between 1.5 and 2.5 ppm. The chemical shift at 3.7 ppm is the characteristic peak of the protons of –CH2Cl of the CEMA segment. The characteristic peak of the protons of –CH3 of DEVP unit is located at 1.3 ppm. The chemical shifts in the region of 3.9–4.6 ppm are assigned to the protons of –OCH2– of both CEMA and DEVP units. Three protons of the methyl group of CEMA unit generate a broad doublet in 0.8–1.3 ppm range, which indicates partial isotacticity of the PCEMA structure.
A typical MALDI–ToF mass spectrum of P(DEVP-co-CEMA) (Sample F3 in Table ) is shown in Figure , in which the dithranol (matrix) and the reflection mode are selected for the polymer. Five distinct peaks (A–E) are observed within each repeat signal group. The m/z spacings of 148 between the peak A and A1, B and B1, C and C1, D and D1, E and E1 in the spectrum are equal to the mass of CEMA unit. The adjacent peaks (between A and B, B and C, C and D, D and E) are separated by the mass of 16, which are attributed to the mass difference between DEVP unit and CEMA unit. The detailed assignments of each peak are summarized in Table . The analysis result of MALDI–ToF mass spectrum indicates that the low weight fractions are random copolymer, namely DEVP and CEMA units are randomly distributed in the copolymer chains.
Figure 5. The MALDI–ToF mass spectrum of Sample F3 in Table .
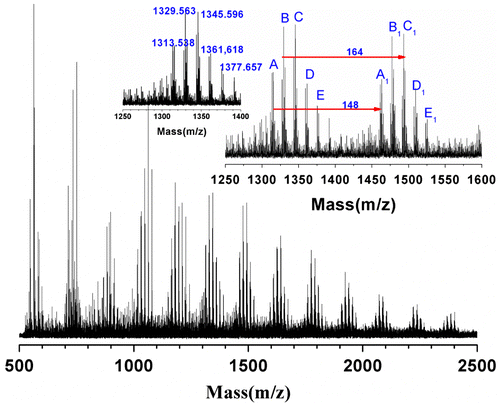
Table 3. Structural assignments of ions appearing in the MALDI–TOF mass spectra of P(DEVP-co-CEMA) (Sample F3 in Table ).
Thermal properties
DSC analyses of P(DEVP-co-CEMA) copolymers (Sample F1, Table ) have been performed compared with the blend of PCEMA and PDEVP on thermal behavior. The low MW fractionated copolymer presents an intermediary Tg of 38.2 °C (Figure (B)), which is between the Tg of PCEMA (95 °C, Mn = 67.0 kDa) and PDEVP (−3 °C, Mn = 107.3 kDa (Figure S21), obtained from coordinative-anionic polymerization, and the synthetic procedure was described in Supplementary Information.) (Figure (C)). The incorporation of DEVP units into the chain of PCEMA lowers the Tg value of PCEMA due to the steric hindrance of phosphonate group. Two Tgs appear clearly in the DSC profile of the blend of PCEMA and PDEVP, which imply the poor compatibility between PCEMA and PDEVP in the blend (Figure (C)). Copolymers fractionated (Sample F1–F7) show lower Tgs (28–54 °C) as compared to the products precipitated directly in hexane (Tables and ), which may attribute to the low MW and narrow molecular weight distribution.
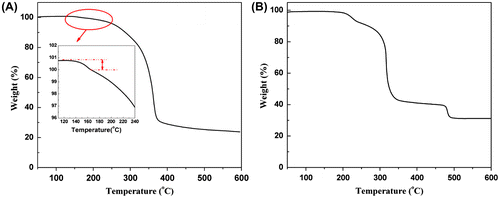
TGA profile of P(DEVP-co-CEMA) copolymer (Figure (A)) exhibits a two-step degradation which is quite different from the blend of PCEMA and PDEVP (which shows a three-step degradation) (Figure (B)). The first step degradation happens because of the side-chain cleavage for PDEVP according to the theory of Bernhard Rieger.[Citation30] The second degradation step is presumably caused by the main-chain scission of both CEMA and DEVP units. The main decomposition step occurs at 260–370 °C.
Figure 6. TGA profiles of Sample F3 (A) and the blend of PCEMA and PDEVP (B).
Self-assembly behavior of amphiphilic random copolymers
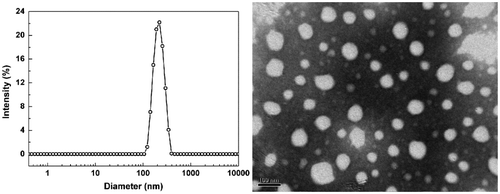
P(DEVP-co-CEMA) is a class of amphiphilic copolymers, so the self-assembly behavior of the copolymers in aqueous solution is investigated. The copolymer aggregates are obtained by adding deionized water dropwise, as a precipitant, to the solutions of P(DEVP-co-CEMA) in THF and then followed by dialysis against deionized water to remove the organic solvent. DLS analyses demonstrate that the sample F3 forms nanoparticles with a size of 207 nm (PDI = 0.036) in aqueous solution (Figure ). TEM image indicates that regular spherical nanoparticles are formed in water with a diameter of 50 to 200 nm (Figure ). Sample F6 forms nanoparticles with a diameter of 203 nm (PDI = 0.05) in aqueous solution (Figure S22). The size of the nanoparticles seems to vary little with the tuning of copolymer composition, which may relate to the random structure of the copolymers.
Figure 7. TEM image and the corresponding DLS result of P(DEVP-co-CEMA) (Sample F3 in Table ) nanoparticles in water.
Scheme 1. A representative reaction procedure for the synthesis of P(DEVP-co-CEMA) copolymers via conventional free radical polymerization.
Conclusions
P(DEVP-co-CEMA) copolymers are successfully synthesized by conventional free radical copolymerization. The amphiphilic copolymers with relatively narrow MW distribution and higher DEVP content are isolated and purified from the reaction mixtures through precipitation fractionation. MALDI–ToF and DSC measurements demonstrate the random structure of the resultant copolymers. The copolymers exhibit a two-step degradation, and the main degradation occurs at about 300 °C. Furthermore, P(DEVP-co-CEMA) copolymers can self-assemble into nanoparticles in aqueous solution for their amphiphilic properties. P(DEVP-co-CEMA) copolymers are of special interest not only for being a class of phosphorus-containing copolymers, but also for the presence of the chlorine atom in the side chain. They can either serve as a macroinitiator for ATRP or become azido functionalities for click chemistry. The modified copolymers show great potential in biomedical fields and flame retardant area.
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.10.1080/15685551.2015.1041078.
Acknowledgments
The authors gratefully acknowledge the financial supports of the National Natural Science Foundation of China (21174121), the Special Funds for Major Basic Research Projects (G2011CB606001), and Zhejiang Provincial Natural Science Foundation of China (LY13B040001).
Additional information
Funding
References
- Parvole J, Jannasch P. Polysulfones grafted with poly(vinylphosphonic acid) for highly proton conducting fuel cell membranes in the hydrated and nominally dry state. Macromolecules. 2008;41:3893–3903.10.1021/ma800042m
- Schuster M, Rager T, Noda A, Kreuer KD, Maier J. About the choice of the protogenic group in pem separator materials for intermediate temperature, low humidity operation: a critical comparison of sulfonic acid, phosphonic acid and imidazole functionalized model compounds. Fuel Cells. 2005;5:355–365.10.1002/(ISSN)1615-6854
- Steininger H, Schuster M, Kreuer KD, Kaltbeitzel A, Bingol B, Meyer WH, Schauff S, Brunklaus G, Maier J, Spiess HW. Intermediate temperature proton conductors for PEM fuel cells based on phosphonic acid as protogenic group: a progress report. Phys. Chem. Chem. Phys. 2007;9:1764–1773.10.1039/b618686f
- Souquet-Grumey J, Perrin R, Cellier J, Bigarre J, Buvat P. Synthesis and fuel cell performance of phosphonated hybrid membranes for PEMFC applications. J. Membr. Sci. 2014;466:200–210.10.1016/j.memsci.2014.04.006
- Berber MR, Fujigaya T, Nakashima N. High-temperature polymer electrolyte fuel cell using poly(vinylphosphonic acid) as an electrolyte shows a remarkable durability. Chem. Cat. Chem. 2014;6:567–571.10.1002/cctc.v6.2
- Berber MR, Fujigaya T, Sasaki K, Nakashima N. Remarkably durable high temperature polymer electrolyte fuel cell based on poly(vinylphosphonic acid)-doped polybenzimidazole. Sci. Rep. 2013;3:1764–1770.
- Lee WL, Liu LC, Chen CM, Lin JS. Syntheses and flame retarding properties of DOPO polymers, melamine polymers, and DOPO-melamine copolymers. Polym. Adv. Technol. 2014;25:36–40.10.1002/pat.v25.1
- Wang TL, Cho YL, Kuo PL. Flame-retarding materials. II. Synthesis and flame-retarding properties of phosphorus-on-pendent and phosphorus-on-skeleton polyols and the corresponding polyurethanes. J. Appl. Polym. Sci. 2001;82:343–357.10.1002/(ISSN)1097-4628
- Ménard R, Negrell-Guirao C, Ferry L, Sonnier R, David G. Synthesis of new flame-retardants by radical chain transfer copolymerization of glycidyl methacrylate and dimethoxy-phosphorylmethyl methacrylate. Eur. Polym. J. 2014;57:109–120.10.1016/j.eurpolymj.2014.05.006
- Carja ID, Serbezeanu D, Lisa G, Vlad-Bubulac T, Hamciuc C. Thermal degradation and kinetic studies of new flame-retardant phosphorus-containing polymers with liquid crystalline properties. Int. J. Polym. Anal. Charact. 2014;19:372–382.10.1080/1023666X.2014.902896
- Monge S, Canniccioni B, Graillot A, Robin JJ. Phosphorus-containing polymers: a great opportunity for the biomedical field. Biomacromolecules. 2011;12:1973–1982.10.1021/bm2004803
- Abueva CDG, Lee BT. Poly(vinylphosphonic acid) immobilized on chitosan: a glycosaminoglycan-inspired matrix for bone regeneration. Int. J. Biol. Macromol. 2014;64:294–301.10.1016/j.ijbiomac.2013.12.018
- Westheimer FH. Why nature chose phosphates. Science. 1987;235:1173–1178.
- Palmer CD, Ninković J, Prokopowicz ZM, Mancuso CJ, Marin A, Andrianov AK, Dowling DJ, Levy O. The effect of stable macromolecular complexes of ionic polyphosphazene on HIV Gag antigen and on activation of human dendritic cells and presentation to T-cells. Biomaterials. 2014;35:8876–8886.10.1016/j.biomaterials.2014.06.043
- Ucan D, Kanik FE, Karatas Y, Toppare L. Synthesis and characterization of a novel polyphosphazene and its application to biosensor in combination with a conducting polymer. Sens. Actuators B. 2014;201:545–554.10.1016/j.snb.2014.05.040
- Steinbach T, Alexandrino EM, Wahlen C, Landfester K, Wurm FR. Poly(phosphonate)s via Olefin Metathesis: adjusting hydrophobicity and morphology. Macromolecules. 2014;47:4884–4893.10.1021/ma5013286
- David G, Negrell-Guirao C, Iftene F, Boutevin B, Chougrani K. Recent progress on phosphonate vinyl monomers and polymers therefore obtained by radical (co)polymerization. Polym. Chem. 2012;3:265–274.10.1039/C1PY00276G
- Ford-Moore AH, Howarth Williams J. The reaction between trialkyl phosphites and alkyl halides. J. Chem. Soc. 1947;1465–1467.10.1039/jr9470001465
- Kosolapoff GM. Isomerization of tri-alkyl phosphites. II. Reaction between triethyl phosphite and trimethylene bromide. J. Am. Chem. Soc. 1944;66:1511–1512.10.1021/ja01237a028
- Kosolapoff GM. Isomerization of tri-alkyl phosphites. J. Am. Chem. Soc. 1944;66:109–111.10.1021/ja01229a031
- Kosolapoff GM. Isomerization of alkyl phosphites. VII. Some derivatives of 2-bromoethane phosphonic acid. J. Am. Chem. Soc. 1948;70:1971–1972.10.1021/ja01185a512
- Pike RM, Cohen RA. Organophosphorus polymers. I. Peroxide-initiated polymerization of diethyl and diisopropyl vinylphosphonate. J. Polym. Sci. 1960;44:531–538.10.1002/pol.1960.1204414424
- Bingöl B, Hart-Smith G, Barner-Kowollik C, Wegner G. Characterization of oligo(vinyl phosphonate)s by high-resolution electrospray ionization mass spectrometry: implications for the mechanism of polymerization. Macromolecules. 2008;41:1634–1639.10.1021/ma702225k
- Sato T, Hasegawa M, Seno M, Hirano T. Radical polymerization behavior of dimethyl vinylphosphonate: homopolymerization and copolymerization with trimethoxyvinylsilane. J. Appl. Polym. Sci. 2008;109:3746–3752.10.1002/app.v109:6
- Kawauchi T, Ohara M, Udo M, Kawauchi M, Takeichi T. Preparation of isotactic-rich poly(dimethyl vinylphosphonate) and poly(vinylphosphonic acid) via the anionic polymerization of dimethyl vinylphosphonate. J. Polym. Sci. Polym. Chem. 2010;48:1677–1682.10.1002/pola.v48:8
- Perrin R, Elomaa M, Jannasch P. Nanostructured proton conducting polystyrene−poly(vinylphosphonic acid) block copolymers prepared via sequential anionic polymerizations. Macromolecules. 2009;42:5146–5154.10.1021/ma900703j
- Wagner T, Manhart A, Deniz N, Kaltbeitzel A, Wagner M, Brunklaus G, Meyer WH. Vinylphosphonic acid homo- and block copolymers. Macromol. Chem. Phys. 2009;210:1903–1914.10.1002/macp.v210:22
- Li J, Ni XF, Ling J, Shen ZQ. Syntheses and properties of poly(diethyl vinylphosphonate) initiated by lanthanide tris(borohydride) complexes: polymerization controllability and mechanism. J. Polym. Sci. Polym. Chem. 2013;51:2409–2415.10.1002/pola.26626
- Salzinger S, Rieger B. Rare earth metal-mediated group transfer polymerization of vinylphosphonates. Macromol. Rapid Commun. 2012;33:1327–1345.10.1002/marc.201200278
- Salzinger S, Seemann UB, Plikhta A, Rieger B. Poly(vinylphosphonate)s synthesized by trivalent cyclopentadienyl lanthanide-induced group transfer polymerization. Macromolecules. 2011;44:5920–5927.10.1021/ma200752d
- Salzinger S, Soller BS, Plikhta A, Seemann UB, Herdtweck E, Rieger B. Mechanistic studies on initiation and propagation of rare earth metal-mediated group transfer polymerization of vinylphosphonates. J. Am. Chem. Soc. 2013;135:13030–13040.10.1021/ja404457f
- Zhang N, Salzinger S, Rieger B. Poly(vinylphosphonate)s with widely tunable LCST: a promising alternative to conventional thermoresponsive polymers. Macromolecules. 2012;45:9751–9758.10.1021/ma3019014
- Zhang N, Salzinger S, Soller BS, Rieger B. Rare earth metal-mediated group-transfer polymerization: from defined polymer microstructures to high-precision nano-scaled objects. J. Am. Chem. Soc. 2013;135:8810–8813.10.1021/ja4036175
- Yang JM, Liang YJ, Salzinger S, Zhang N, Dong DW, Rieger B. Poly(vinylphosphonate)s functionalized polymer microspheres via rare earth metal-mediated group transfer polymerization. J. Polym. Sci. Polym. Chem. 2014;52:2919–2925.10.1002/pola.27324
- Rabe GW, Komber H, Häussler L, Kreger K, Lattermann G. Polymerization of diethyl vinylphosphonate mediated by rare-earth tris(amide) compounds. Macromolecules. 2010;43:1178–1181.10.1021/ma902068n
- Seemann UB, Dengler JE, Rieger B. High-molecular-weight poly(vinylphosphonate)s by single-component living polymerization initiated by rare-earth-metal complexes. Angew. Chem. Int. Ed. 2010;49:3489–3491.10.1002/anie.201000804
- Kehrle J, Hohlein IMD, Yang ZY, Jochem AR, Helbich T, Kraus T, Veinot JGC, Rieger B. Thermoresponsive and photoluminescent hybrid silicon nanoparticles by surface-initiated group transfer polymerization of diethyl vinylphosphonate. Angew. Chem. Int. Ed. 2014;53:12494–12497.
- Soller BS, Zhang N, Rieger B. Catalytic precision polymerization: rare earth metal-mediated synthesis of homopolymers, block copolymers, and polymer brushes. Macromol. Chem. Phys. 2014;215:1946–1962.10.1002/macp.v215.20
- Komber H, Steinert V, Voit B. 1H, 13C, and 31P NMR study on poly(vinylphosphonic acid) and its dimethyl ester. Macromolecules. 2008;41:2119–2125.10.1021/ma702662q
- Bingöl B, Meyer WH, Wagner M, Wegner G. Synthesis, microstructure, and acidity of poly(vinylphosphonic acid). Macromol. Rapid Commun. 2006;27:1719–1724.10.1002/(ISSN)1521-3927
- Li XS, Xu LD, Shan YB, Yuan BF, Feng YQ. Preparation of magnetic poly(diethyl vinylphosphonate-co-ethylene glycol dimethacrylate) for the determination of chlorophenols in water samples. J. Chromatogr. A. 2012;1265:24–30.10.1016/j.chroma.2012.09.083
- Arcus CL, Matthews RJS. 883. Phosphonic polymers. Part I. The copolymerisation of diethyl vinylphosphonate and styrene. J. Chem. Soc. 1956;4607–4612.10.1039/jr9560004607
- Lindsey RV; E. I. Du Pont de Nemours & Co. Copolymer of α,β-ethylenically unsaturated phosphonic acid derivatives. United States patent US 2,439,214. 1948 Apr 6.
- Banks M, Ebdon JR, Johnson M. The flame-retardant effect of diethyl vinyl phosphonate in copolymers with styrene, methyl-methacrylate, acrylonitrile and acrylamide. Polymer. 1994;35:3470–3473.10.1016/0032-3861(94)90910-5
- Gunes D, Karagoz B, Bicak N. Synthesis of methacrylate-based functional monomers via boron ester acidolysis and their polymerization. Des. Monomers Polym. 2009;12:445–454.10.1163/138577209X12486896623571
- Babu GN, Narula A, Lu PH, Li X, Hsu SL, Chien JCW. Radiolysis of resist polymers. 2. Poly(haloalkylmethacrylates) and copolymers with methyl methacrylate. Macromolecules. 1984;17:2756–2761.10.1021/ma00142a055
- Babu GN, Chien JCW. Radiolysis of resist polymers. 5. Poly(haloalkyl α-chloroacrylates) and copolymers with methyl methacrylate. Macromolecules. 1984;17:2761–2764.10.1021/ma00142a056
- Ding JX, Xiao CS, He CL, Li MQ, Li D, Zhuang XL, Chen XS. Facile preparation of a cationic poly(amino acid) vesicle for potential drug and gene co-delivery. Nanotechnology. 2011;22:494012–494020.
- Li MM, Shan GR, Bao YZ, Pan PJ. Poly(ε-caprolactone)-graft-poly(N-isopropylacrylamide) amphiphilic copolymers prepared by a combination of ring-opening polymerization and atom transfer radical polymerization: synthesis, self-assembly, and thermoresponsive property. J. Appl. Polym. Sci. 2014;131:41115–41123.
- Zhang Y, Zhao J, Yang P, He SJ, Huang HJ. Synthesis and characterization of energetic GAP-b-PAEMA block copolymer. Polym. Eng. Sci. 2012;52:768–773.10.1002/pen.v52.4
- Price D, Pyrah K, Hull TR, Milnes GJ, Ebdon JR, Hunt BJ, Joseph P, Konkel CS. Flame retarding poly(methyl methacrylate) with phosphorus-containing compounds: comparison of an additive with a reactive approach. Polym. Degrad. Stab. 2001;74:441–447.10.1016/S0141-3910(01)00184-7
- Blunden BM, Thomas DS, Stenzel MH. Macromolecular ruthenium complexes as anti-cancer agents. Polym. Chem. 2012;3:2964–2975.10.1039/c2py20439h
- Fineman M, Ross SD. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950;5:259–262.10.1002/pol.1950.120050210