
Abstract
Polycarbosilanes were prepared via hydrosilylation polymerization of functional 1,1-diphenylethylene (DPE) derivatives (dimethyl-[4-(1-phenylvinyl)phenyl]silane (DPE-SiH) and 1-(4-methyloxyphenyl)-1′-(4-dimethylsilanephenyl) ethylene (DPE-SiH/OMe)). Both DPE-SiH and DPE-SiH/OMe were synthesized as monomers and then utilized to prepare corresponding oligomers with Si–C bonds and phenyl rings in their backbones. The DPs were calculated to be 5.9 and 6.3, respectively, while the β-additions were 88.8 and 88.0%. The Td5 of DPE-SiH and DPE-SiH/OMe oligomers were 270 and 345 °C, respectively, and their weight residue was approximately 20% even heated at a temperature near 1200 °C in nitrogen. The compact tiny pores distributed on cross section of the resultant SiC ceramics were observed with the scanning electron microscopy. The thermal analyses verified that the oligomers of DPE-SiH and DPE-SiH/OMe possessed favorable thermal stabilities and could be taken as the proper PCSs precursor for preparation of SiC ceramic materials.
1. Introduction
The polycarbosilanes (PCSs) with Si–C bonds in their backbone chains are important materials in the fields of aerospace, electrical appliance, and mechanology for their special properties.[Citation1] Additionally, in past few decades, it has been found that PCSs can serve as organosilicon precursor to prepare SiC- or Si/C/N-based ceramic materials,[Citation2] and offer SiC ceramic material the attractive polymer forming capabilities and outstanding properties such as high-temperature stability, high strength, and corrosion resistance. In the case of taking PCSs as precursors, the resultant SiC ceramic and fiber materials can be advantageously manufactured by cementation and spinning.[Citation3] Meanwhile, the C/Si ratios, structures, and functions of the SiC ceramic materials can be adjusted diversely through molecular design and selection in preparation of PCSs precursors. Furthermore, the incorporation of the functional or rigid groups into the backbone chain can be smoothly realized with the method mentioned above, and therefore, the modified SiC ceramic materials can be acquired as desired.
The hydrosilylation is actually a kind of coupling reaction between Si–H bonds and alkene or alkyne and is featured of high efficiency and wide application range under mild reaction conditions. Diverse functional moieties can be introduced into polymer chains conveniently by dint of hydrosilylation strategies, and the targeted in-chain or chain-end functional polymers can be prepared efficiently.[Citation4,5] As derived from the hydrosilylation, hydrosilylation polymerization is a step-wise polymerization method for synthesizing polymers with Si–C bonds in their backbone chains from the multi-functional organosilicon. Thus, hydrosilylation polymerization is deemed as a versatile synthetic methodology to prepare saturated organosilicon polymers. Intermolecular hydrosilylation polymerization is a significant method rooted in hydrosilylation polymerizations [Citation6] because the structure of the polymer to be prepared is controllable and the monomer is relatively apt to obtain; the only restriction in this reaction is that the monomers must contain both alkenyl/alkynyl groups and Si–H bonds in a single molecule. Furthermore, the resultant polycarbosilanes are endued with outstanding solubility and high thermal stability after introduction of phenyl rings in the side groups because the atom of chiral center in the main chain can decrease the crystallinity.[Citation7]
More recently, 1,1-diphenylethylene (DPE) and its derivatives have been widely used for quantitative synthesis of the in-chain and chain-end functionalized polymers.[Citation8] Because its special structure has remarkable steric hindrance, DPE cannot be homopolymerized with conventional chain growth polymerization method. Furthermore, functional groups can be introduced into dual phenyl rings of DPE conveniently. Considering the superiority of hydrosilylation, dimethyl-[4-(1-phenylvinyl) phenyl]silane (DPE-SiH) is synthesized and used to copolymerization. Subsequently, Quirk et al. [Citation9] proposed the general anionic functionalization methods (GFM) to enrich the base polymers for post-functionalization.[Citation10] A variety of functional groups can be linked to polymer chains efficiently and conveniently by means of GFM. As the core of GFM, DPE-SiH is used to not only synthesize the chain-end Si–H functionalized polymers,[Citation11,12] but also copolymerize with styrene/butadiene/isoprene to synthesize the in-chain multi-functionalized copolymers.[Citation13,14] In addition, functional moieties can be introduced into the polymers through hydrosilylation.[Citation11–13] However, the polymer with further functionalized DPE-SiH derivatives (i.e. containing another functional group besides Si–H) as monomer have not been prepared anywhere. Meanwhile, as a functional monomer containing Si–H and vinyl groups simultaneously, DPE-SiH and its derivatives can also be utilized for further functionalized reactions.
2. Experimental
2.1. Materials
Tetrahydrofuran (THF) was dried by reflux over the Na-benzophenone complex under the argon atmosphere and then distilled. Dimethylchlorsilane (Aldrich, 98%) was dried by reflux over CaH2 under the argon atmosphere and then distilled. Methyltriphenylphosphonium bromide (Energy Chemical, 98%), potassium tert-butoxide (Energy Chemical, 98%), magnesium turnings for Grignards (J&K, 99.8%), (4-bromophenyl)(4′-methoxyphenyl)methanone (BP-Br/OMe) (Novochemy, 97%), and Karstedt catalyst (Aldrich, 98%) were used as received. Other reagents were purchased from Sinopharm Chemical Reagent Company and used as received unless otherwise stated.
2.2. Measurements
1H NMR (5 wt%, CDCl3) spectra were recorded on a Bruker Avancell 400 M NMR spectrometer with tetramethylsilane as the internal standard. Size exclusion chromatographic (SEC) analyses of the copolymers were performed on a Waters HPLC component system (2414 refractive index detector) at a flow rate of 1.0 mL/min in THF at 30 °C after calibrated using polystyrene standard polymers. Glass transition temperatures (Tg) of the polymers were measured under nitrogen atmosphere using a TA Instruments Universal Analysis 2000 differential scanning calorimeter (DSC) at a heating rate of 10.0 °C/min, and sample sizes were 5 mg and investigated under nitrogen. The TGA analysis were performed under nitrogen atmosphere (heating rate 20.0 °C/min, nitrogen flow 100 mL/min) using SDT (Simultaneous TGA/DSC) Q600 (TA Instruments). The mass of the sample used was typically 10.0 mg, and samples were placed in Al2O3 crucible and weighed directly. Scanning electronic microscopy (SEM) observations were carried out using a FEI Quanta 200F at an acceleration voltage of 20 kV. Sample surfaces were sputtered a layer of gold before SEM observation.
2.3. Synthesis
4-bromodiphenylethylene (DPE-Br) and dimethyl-[4-(1-phenylvinyl) phenyl]silane (DPE-SiH) were prepared as in the literature.[Citation12] The synthetic routes for monomers and oligomers are shown in Figure (a).
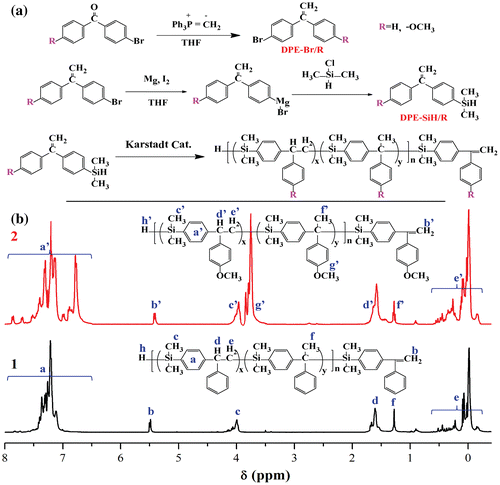
Figure 1. (a) The synthetic routes of monomers and polycarbosilane oligomers; x and y are the contents of β-addition and α-addition in corresponding oligomers, respectively and (b) the 1H NMR spectra of polycarbosilane oligomers 1 and 2.
2.3.1. 1-(4-bromophenyl)-1′-(4-methoxyphenyl)ethylene (DPE-Br/OMe)
A cooled (ice bath, 0 °C) 1000 mL three-necked round-bottomed flask was equipped with a magnetic stirring bar, and methyltriphenylphosphonium bromide (62.01 g, 0.174 mol) and dry THF (100 mL) was added successively and stirred. Potassium tert-butoxide (17.95 g, 0.16 mol) was dissolved in 100 mL THF and added dropwise into the flask at 0 °C under nitrogen atmosphere. After stirred half an hour, BP-Br/OMe (29.1 g, 0.1 mol) was dissolved in 300 mL THF and drop-wise added into the flask. The reaction was stirred under a nitrogen atmosphere for 12 h, and 10 mL H2O was added into the flask to terminate the reaction. The mixture was diluted with brine and exacted with diethyl ether for three times (3 × 200 mL), and then, the aqueous layer was further extracted with diethyl ether, and the organic layer was collected and dried with anhydrous MgSO4 overnight. The resolution was filtrated and concentrated under reduced pressure, and then, the flash column chromatography was taken as twice using hexane (Rf = 0.46). The 91% yield of DPE-Br/OMe (26.3 g, 0.091 mol) was reserved, and it was characterized by 1H NMR (CDCl3): δ 5.33, 5.39 (d, 2H,=CH2), 3.80 (s, 3H, O–CH3), 6.86, 7.23, 7.45 ppm (m, 8H, aromatic C–H).
2.3.2. 1-(4-methyloxyphenyl)-1′-(4-dimethylsilanephenyl)ethylene (DPE-SiH/OMe)
A grain of iodine crystal and 12 g magnesium shavings (0.5 mol) were added into a three-necked round-bottomed flask. DPE-Br/OMe (28.9 g, 0.1 mol) was dissolved in 600 mL dried THF and drop-wise added into the flask. Then, the flask was heated to THF reflux and kept for four hours under a nitrogen atmosphere. Until the Grignard reaction completed, the solution was transferred to another three-necked flask. Then, chlorodimethylsilane (16.7 mL, 0.15 mol) was drop-wise added into the same flask, and the solution was stirred at room temperature overnight. Then, the reaction was terminated by adding deionized water, the mixture was diluted with brine and exacted with diethyl ether for 3 times (3 × 200 mL), the aqueous layer was further extracted with diethyl ether, and the organic layer was collected and dried over anhydrous MgSO4 overnight. The resolution was filtrated and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexane/ethyl acetate 29:1 v/v (Rf = 0.6) followed by re-crystallization twice to give pure DPE-SiH/OMe in 78% yield (20.9 g, 0.078 mol). 1H NMR (CDCl3): δ 0.36 (s, 6H, Si(CH3)2), 3.82 (s, 3H, O–CH3), 4.44 (m, 1H, Si–H), 5.37, 5.41(d, 2H, =CH2), 6.86, 7.27, 7.33 ppm (m, 8H, aromatic C–H). 13C NMR (CDCl3): δ −3.57 (Si–(CH3)2), 55.31 (O–CH3), 113.5 (C=C*H2), 136.85 (aromatic C*-Si(CH3)2H), and 149.59 ppm (>C*=CH2). 29Si NMR (CDCl3): δ −17.446 ppm (aromatic C–Si*(CH3)2H).
2.4. Hydrosilylation polymerization
Taken P(DPE-SiH) as an example for illustration of preparation, 4.76 g (0.02 mol) DPE-SiH monomer was put into argon-protected Schlenk flask, kept stirring, and then, 0.017 mmol Karstedt catalyst was added and the reactant mixture was heated up to 65 °C and stirred for 240 h. As the polymerization degree increased, the viscosity of the reaction system raised up. And 5 mL toluene was added into the flask when the oligomer became hard to stir. The polymerization solution was precipitated in methanol after the reaction was terminated. Then, the sediment was refined by aluminum oxide 150 basic column chromatography using ethyl acetate: n-hexane = 1:9 and pure ethyl acetate sequentially, and dried to constant weight.
2.5. The kinetic study
Take II as an example for the illustration: 0.02 mol DPE-SiH/OMe and 0.017 mmol Karstedt catalyst was put into a Schlenk flask and stirred at 65 °C for 240 h. During the reaction, 5 samples of 0.5 mL solution were taken from this system every 48 h. Each sample was disposed as the Section 2.4, and then, all the oligomers were characterized by 1H NMR after the unreacted monomer was removed, and the results are shown in Figure and Table .
Figure 2. The kinetic curves of DPE-SiH/OMe hydrosilylation polymerization (a) Dependence of ln([SiH]0/[SiH]) on the reaction time and (b) DP on the reaction time.
![Figure 2. The kinetic curves of DPE-SiH/OMe hydrosilylation polymerization (a) Dependence of ln([SiH]0/[SiH]) on the reaction time and (b) DP on the reaction time.](/cms/asset/16a56047-5212-4bfa-a6b5-b95ba94d596a/tdmp_a_1041079_f0002_oc.gif)
3. Results and discussion
DPE could not be homopolymerized by ‘chain propagation’ because of the existence of its steric hindrance.[Citation15] In this work, the syntheses of DPE-SiH and DPE-SiH/OMe oligomers were carried out through hydrosilylation polymerization. The hydrosilylation of DPE-SiH or DPE-SiH/OMe were performed at 65 °C for 720 h. Their corresponding oligomers were obtained and characterized; the results are listed in Table .
Table 1. The characteristics of DPE-SiH and DPE-SiH/OMe oligomers obtained by hydrosilylation polymerization.
From the previous studies, it was found that two regioisomers (α-addition/β-addition) [Citation16] were usually generated in the presence of platinum catalysts. In the 1H NMR spectra presented in Figure (b), c and c′ were characteristic peaks of methine groups which were produced by β-addition; d and d′ were characteristic peaks of methylene groups which were originated from β-addition; and f and f′ were characteristic peaks of methyl groups as the result of α-addition. The proportion of β-addition was 88.8% for P(DPE-SiH) and 88.0% for P(DPE-SiH/OMe), as calculated from Equation (1) according to the area integrals.(1)
The terminal Si–H peaks (h and h′) of the oligomers cannot be observed in the spectra of 1H NMR (Figure (b)), which may be because the terminal Si–H groups has been hydrolyzed with the hydroxyl groups of solvents (here is the methanol which was used to precipitate oligomers) under high concentrations of platinum catalysts.[Citation17–19]
The effect of temperature and catalyst concentration on hydrosilylation polymerization of DPE-SiH/OMe was investigated. The kinetic curves are shown in Figure (a). The first-order rate constants, k1, were obtained from the slopes of ,[Citation20] and are listed in Table .
Table 2. The kinetic results of DPE-SiH/OMe hydrosilylation polymerization.
Linear kinetic curves were obtained from both vs. Time and DP vs. Time (in Figure (a) and (b)); it could be observed that the hydrosilylation polymerization of DPE-SiH/OMe exhibited typical behavior of a step-wise polymerization.[Citation21] At 100 °C, I was obtained with low DP due to precipitation of platinum catalyst (insoluble black particle was obvious in the bottom of flask, which has been reported in previous work.[Citation22]) The reaction rate constant k1 for I was relatively lower than that of II in the DP vs. Time curve. The polymerization rate of III was relatively high due to the highest concentration of Karstedt catalyst; however, the catalyst produced excess active center and excess side reaction. Thus, the DP of III showed a great fluctuation and lower than II at the end. To contrast the product 2 in Table with the product II in Table , it could be found that the DP of P(DPE-SiH/OMe) was 5.2 in 240 h and raised up to 6.3 in 720 h. As reaction time prolonged 3 times, the DP increased scarcely although monomer was excess.
Thermal properties of these two oligomers were investigated by DSC and TGA (Table and Figure ). The Tg was about 20 °C, because of the relatively low DP. Substituent groups had certain effect on polymer’s thermal performance: Tg of oligomer 2 was slightly higher than that oligomer 1, since the latter had no methoxy substituent groups in similar DP.
Figure 3. The TGA curves of polycarbosilane oligomers 1 and 2 under nitrogen stream.
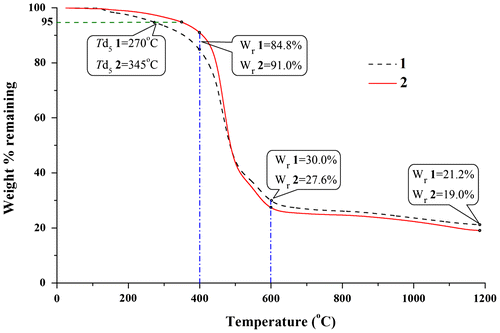
As it can be seen from the TGA curves in Figure , the methoxy groups appeared to have important effect on thermal stability, evidenced by oligomer 1’s Td5 of 270 °C and oligomer 2’s Td5 of 345 °C; oligomer 2 exhibited only a 9% weight loss, while oligomer 1 lost 15% when heated to 400 °C under nitrogen atmosphere. However, when the samples were heated above 500 °C, the weight residue of oligomer 1 and 2 was approximately equal in value. The TGA curves tended to level off steadily due to H, O, and redundant C composition nearly decomposed completely when the samples were heated above 600 °C; the weight residues were approximate 20% when they were heated at near 1200 °C in nitrogen. The residues of the two oligomers were characterized by SEM, and the micrographs obtained are shown in Figure . As observed in SEM images, compact tiny pores could be seen on cross section of the resultant SiC ceramics. The thermal analyses verified that the oligomers of DPE-SiH and DPE-SiH/OMe possessed favorable thermal stabilities and could act as proper PCSs precursor for preparation of SiC ceramic materials.
Figure 4. SEM images of SiC Ceramic cross section sintered form polycarbosilane oligomers 1 and 2.

4. Conclusions
Two kinds of 1,1-diphenylethlene derivative monomers (DPE-SiH and DPE-SiH/OMe) containing Si–H groups were successful synthesized in this work. Subsequently, two kinds of oligomers, P(DPE-SiH) and P(DPE-SiH/OMe), were prepared via hydrosilylation polymerization. The DPs of the two oligomers were calculated to be 5.9 and 6.3, respectively, in terms of 1H NMR spectra, and the proportion of their β-addition was 88.8 and 88.0%, respectively. The kinetics of the corresponding polymerizations was investigated using the DPE-SiH/OMe as monomer, through the variation of catalyst content and reaction temperatures. The excess catalyst activity center (SiH:Pt = 600:1) produced the highest k1 (1.58 × 10−2 h−1) at 65 °C, but lower DP was obtained.
Introduction of methoxyl groups could improve oligomer’s thermal stability. Being similar to DPs, Td5 of DPE-SiH/OMe oligomers with methoxyl groups in the chain raised up to 345 °C from 270 °C of DPE-SiH oligomers. Furthermore, the weight residues of both oligomers were approximately 20% when they were heated at near 1200 °C in nitrogen.
At present, novel 1-phenyl-1′-(4-dimethylsilanephenyl)ethylene derivative monomers and corresponding oligomers are being prepared. Meanwhile, the works on improving the DP of PDPE and maximizing its thermal properties are still in progress.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Krska SW, Seyferth D. Synthesis of water-soluble carbosilane dendrimers. J. Am. Chem. Soc. 1998;120:3604–3612.10.1021/ja971865o
- Birot M, Pillot JP, Dunogues J. Comprehensive chemistry of polycarbosilanes, polysilazanes, and polycarbosilazanes as precursors of ceramics. Chem. Rev. 1995;95:1443–1477.10.1021/cr00037a014
- Yajima S, Hasegawa Y, Okamura K, Matsuzawa T. Development of high tensile strength silicon carbide fibre using an organosilicon polymer precursor. Nature. 1978;273:525–527.10.1038/273525a0
- Yu XF, Zhong S, Li XP, Tu YF, Yang SG, Van Horn RM, Ni CY, Pochan DJ, Quirk RP, Wesdemiotis C, Zhang W-B, Cheng SZD. A giant surfactant of polystyrene−(carboxylic acid-functionalized polyhedral oligomeric silsesquioxane) amphiphile with highly stretched polystyrene tails in micellar assemblies. J. Am. Chem. Soc. 2010;132:16741–16744.10.1021/ja1078305
- Han L, Ma HW, Li Y, Wu J, Xu HY, Wang YR. Construction of topological macromolecular side chains packing model: study unique relationship and differences in LC-microstructures and properties of two analogous architectures with well-designed side attachment density. Macromolecules. 2015;48:925–941.10.1021/acs.macromol.5b00101
- Marciniec B. Hydrosilylation. Adv. Silicon Sci. 2009;1:191–204.
- Tsumura M, Iwahara T, Hirose T. Synthesis and properties of polycarbosilanes by hydrosilylation polymerization. Polym. J. 1995;27:1048–1053.10.1295/polymj.27.1048
- Gausepohl H, Oepen S, Knoll K, Schneider M, McKee G, Loth W. Super-polystyrene: a new class of engineering plastics with versatile properties. Des. Monomers Polym. 2000;3:299–315.10.1163/156855500300160258
- Quirk RP, Garcés C, Collins S, Dabney D, Wesdemiotis C, Dudipala V. Quantitative analysis and characterization of the products of the model reaction of n-butyllithium with excess 1,1-diphenylethylene: 1,1-diphenylhexane and 1,1,3,3-tetraphenyloctane. Polymer. 2012;53:2162–2167.10.1016/j.polymer.2012.03.029
- Hsieh HL, Quirk RP. Anionic polymerization. Principles and practical applications. New York (NY): Marcel Dekker; 1996.
- Quirk RP, Sahoo S. Anionic synthesis of α-functionalized polymers by combination of 1,1-diphenylethylene and hydrosilation chemistry. Macromol. Symp. 2013;325–326:77–88.10.1002/masy.201200043
- Zhang W-B, Sun B, Li H, Ren XK, Janoski J, Sahoo S, Dabney DE, Wesdemiotis C, Quirk RP, Cheng SZD. Synthesis of in-chain-functionalized polystyrene-block-poly(dimethylsiloxane) diblock copolymers by anionic polymerization and hydrosilylation using dimethyl-[4-(1-phenylvinyl)phenyl]silane. Macromolecules. 2009;42:7258–7262.10.1021/ma901506d
- Ma HW, Wang QY, Sang W, Han L, Liu PB, Chen J, Li Y, Wang YR. Synthesis of bottlebrush polystyrenes with uniform, alternating, and gradient distributions of brushes via living anionic polymerization and hydrosilylation. Macromol. Rapid Commun. 2015;36:726−732. doi:10.1002/marc.201400660.
- Ding J, Li Y, Shen KH, Wang B, Wang YR. Anionic synthesis of binary random in-chain multi-functionalized poly(styrene/butadiene/isoprene and dimethyl [4-phenylvinyl)-phenyl]silane) (PS-DPESiH, PB-DPESiH, PI-DPESiH) copolymers. Chin. Chem. Lett. 2012;23:749–752.10.1016/j.cclet.2012.03.012
- Yuki H, Murahashi S, Kosai K, Hotta J. Anionic alternative copolymerization. J. Polym. Sci., Part B: Polym. Lett. 1964;2:1121–1125.10.1002/pol.1964.110021210
- Shintani K, Ooi O, Mori A, Kawakami Y. A facile synthesis of hydro- and vinyl-functionalized di- and tetrasiloxanes and polyaddition via hydrosilylation. Polym. Bull. 1996;37:705–710.10.1007/BF00295766
- Safa KD, Sharifi E, Tofangdarzadeh S, Hassanpour A. Facile synthesis of tris(alkoxydimethylsilyl)methane derivatives via alcoholysis under open-flask and mild conditions. Phosphorus, Sulfur Silicon Relat. Elem. 2010;185:2433–2440.10.1080/10426501003685783
- Safa KD, Tofangdarzadeh S, Ayenadeh HH. Reactions of tris(dimethylsilyl)methane and polymers containing Si–H groups with various hydroxy compounds under aerobic and mild conditions. Heteroat. Chem. 2008;19:365–376.10.1002/(ISSN)1098-1071
- Iwahara T, Kusakabe M, Chiba M, Yonezawa K. Analysis of effervescence during hydrosilylation curing. Polym. J. 1993;25:1303–1310.10.1295/polymj.25.1303
- AntiĆ VV, AntiĆ MP, Govedarica MN, DvorniĆ PR. Kinetics and mechanism of the formation of poly[(1,1,3,3-tetramethyldisiloxanyl)ethylene] and poly(methyldecylsiloxane) by hydrosilylation. J. Polym. Sci. Polym. Chem. 2007;45:2246–2258.10.1002/(ISSN)1099-0518
- Dvornic PR, Gerov VV, Govedarica MN. Polymerization by hydrosilation. 2. Preparation and characterization of high-molecular-weight poly (1,1,3,3-tetramethyldisiloxanyl)ethylene from 1,3-dihydridotetramethyldisiloxane and 1,3-divinyltetramethyldisiloxane. Macromolecules. 1994;27:7575–7580.10.1021/ma00104a011
- Itsuno S, Chao D, Ito K. Hydrosilylation polymerization. I. A new approach to the preparation of macromonomers containing silicon atoms. J. Polym. Sci. Polym. Chem. 1993; 31: 287–291.10.1002/pola.1993.080310135