
Abstract
Four polymerizable phosphonic acids bearing urea groups 4a, 4b, 9a, and 9b as well as the 10-(methacryloyloxy)decylphosphonic acid 10 were synthesized in four to six steps. They were characterized by 1H, 13C, and 31P NMR spectroscopy and by high-resolution mass spectroscopy. Contrary to methacrylates 4a and 4b, methacrylamides 9a and 9b were hydrolytically stable in deuterated water/deuterated ethanol solutions. The photopolymerization behavior of acidic monomers 4a, 4b, 9a, 9b, and 10 and their corresponding phosphonates were studied using a differential scanning calorimeter. Homopolymerizations and copolymerizations with N-butyl-N-ethylacrylamide were carried out. The presence of a urea group was found to significantly increase the rate of polymerization. Monomers bearing a phosphonic acid group were more reactive than their corresponding phosphonates. Self-etch adhesives based on monomers 4a, 4b, 9a, and 9b were able to provide a strong bond between a composite and the dental hard tissues (dentin and enamel). The presence of a urea group led to a significant increase in the shear bond strength to both dentin and enamel.
1. Introduction
Self-etch adhesives (SEAs) are, due to their short and reliable application procedure, widely used to achieve a bond between the dental hard tissues (dentin and enamel) and a restorative material (composite).[Citation1–3] A SEA is an aqueous formulation containing an acidic monomer, monofunctional and crosslinking comonomers (mostly methacrylates), cosolvents (ethanol (EtOH), acetone, etc.), and additives (photoinitiator(s), co-initiator(s), stabilizer, filler, etc.). During the application of the SEA, dental tissues are simultaneously demineralized and infiltrated by the monomers of the adhesive. After drying and photopolymerization of the adhesive layer, the restorative material can be put in place. The acidic monomer, which can be a dihydrogen phosphate as well as a carboxylic or phosphonic acid, is the key component of the SEA.[Citation4] In order to be incorporated in SEAs, this monomer has to fulfill a few requirements.[Citation5] It should exhibit good etching properties, sufficient storage stability, and a low oral toxicity. It also has to demonstrate a high rate of homo- or copolymerization with the comonomers of the formulation. Finally, it should be able to strongly interact with the calcium of hydroxyapatite (HAP). Indeed, Van Meerbeek et al. showed that the adhesive performance of a SEA is directly related to the chemical bonding potential of the acidic monomer to HAP.[Citation6–9] According to the ‘adhesion-decalcification’ concept described by Yoshida et al., the ability of a monomer to achieve a strong chemical bond with HAP is correlated to the solubility of its corresponding calcium salt in water (the lower the solubility, the better the adhesion).[Citation7] In order to improve the interaction with HAP, monomers exhibiting strong chelating properties like diphosphonic acids [Citation10–12] or β-ketophosphonic acids [Citation13] were therefore prepared. Recently, Avci et al. described the synthesis of two monofunctional phosphonated urea methacrylates for dental materials.[Citation14] Photopolymerization investigations have shown that those monomers were more reactive than triethylene glycol dimethacrylate (TEGDMA), a crosslinking monomer. Such highly reactive monofunctional methacrylates are therefore potential candidates to replace crosslinking monomers as diluent in some dental formulations. The high reactivity of urea monomers was attributed to a ‘combination of hydrogen abstraction/chain transfer reactions due to the labile hydrogens and hydrogen bonding imparted to the urea linkage’.[Citation14] The positive impact of hydrogen bonding on the rate of polymerization has already been reported in the literature and was attributed to a preorganization of the monomers, resulting in an increase in the propagation rate as well as to mobility restrictions, which lead to a decrease in the termination rate.[Citation15–18] Phosphonic acids bearing urea groups have so far not been reported in the literature. Such compounds might be able to improve the performance of currents SEAs.
In this context, we took an interest in the synthesis of acidic monomers 4a, 4b, 9a, and 9b (Figure ). The incorporation of the urea group should lead to an increase in the monomer reactivity. Moreover, urea derivatives being able to chelate metals, those monomers should exhibit an improved chemical bonding to HAP.[Citation19] The strong hydrogen bonding ability of the urea moiety might also increase the mechanical properties of the adhesive. In this article, the synthesis, photopolymerization behavior, hydrolytic stability, and adhesive properties of monomers 4a, 4b, 9a, and 9b are described. 10-(methacryloyloxy)decylphosphonic acid 10 was also studied in order to assess the influence of the urea group on the polymerization rate and the adhesive properties (Figure ).
Figure 1. Structure of acidic monomers 4a, 4b, 9a, 9b, and 10.
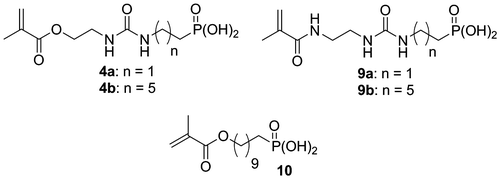
Figure 2. 1H NMR spectrum of acid monomer 4b.
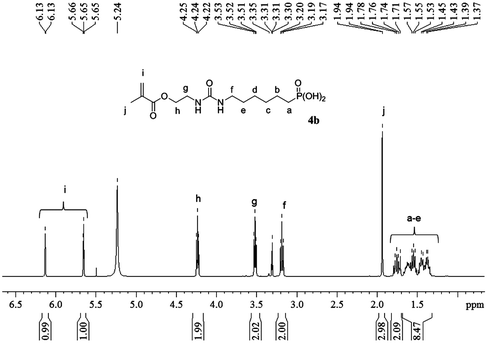
Figure 3. Hydrolysis of acid monomers 4a, 4b, 9a, 9b, and 10.
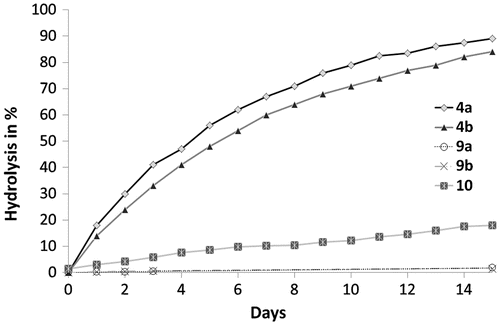
Figure 4. DBC versus irradiation time for the homopolymerization of monomers 3a, 3b, 8a, 8b, HEMA, and TEGDMA.
Figure 5. Rp versus irradiation time for the homopolymerization of monomers 3a, 3b, 8a, 8b, HEMA, and TEGDMA.
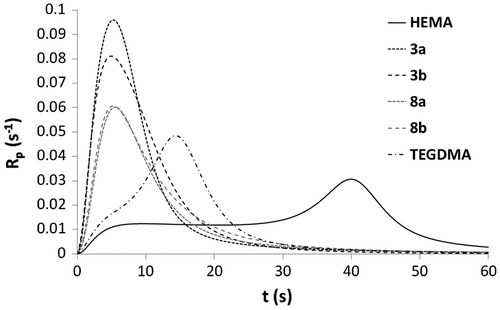
Figure 6. DBC versus irradiation time for the copolymerization of monomers 3a, 3b, 4a, 4b, and 10 with BEA.
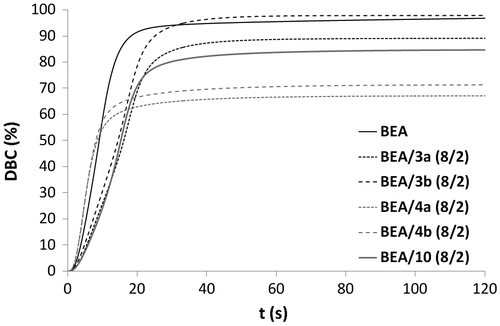
Figure 7. Rp versus irradiation time for the copolymerization of monomers 3a, 3b, 4a, 4b, and 10 with BEA.
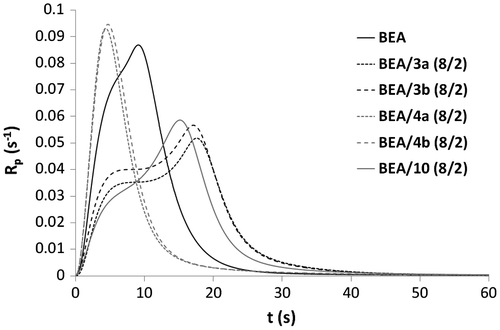
Figure 8. DBC versus irradiation time for the copolymerization of monomer 3b with BEA.
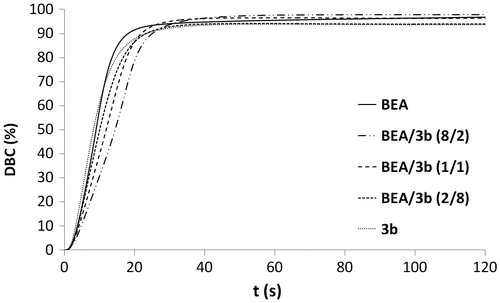
Figure 9. Rp versus irradiation time for the copolymerization of monomer 3b with BEA.
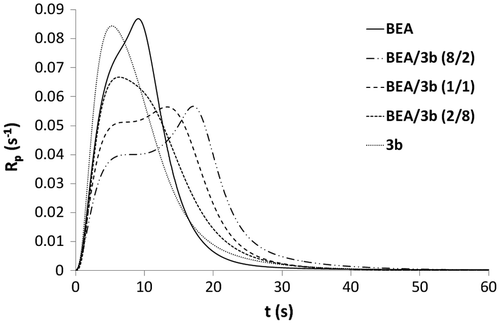
Figure 10. DBC versus irradiation time for the copolymerization of monomers 8a, 8b, 9a, and 9b with BEA.
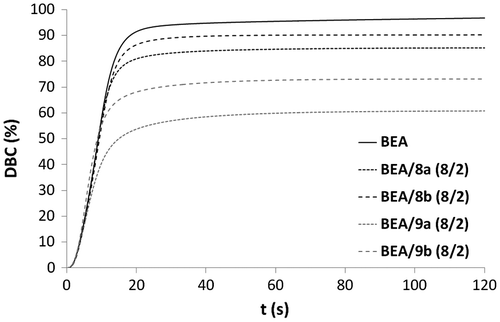
Figure 11. Rp versus irradiation time for the copolymerization of monomers 8a, 8b, 9a, and 9b with BEA.
2. Experimental
2.1. Abbreviations
Ammonium chloride (NH4Cl), bis-(2,4,6-trimethylbenzoyl)phenylphosphinoxide (Irgacure® 819), 2-bis[4-(2-hydroxy-3-methacryloyloxypropoxy)phenyl]propane (Bis-GMA), bis-(4-methoxybenzoyl)diethylgermanium (Ivocerin®), N-butyl-N-ethylacrylamide (BEA), butylated hydroxytoluene (BHT), camphorquinone (CQ), carbodiimidazole (CDI), double-bond conversion (DBC), deuterated ethanol (EtOD6), deuterated water (D2O), dichloromethane (DCM), N,N’-diethyl-1,3-bis-(acrylamido)propane (DEBAAP), 4-dimethylaminopyridine, dimethylformamide (DMF), ethanol (EtOH), ethyl acetate (EA), ethyl 4-(dimethylamino)-benzoate (EMBO), equivalent (eq.), hydroxyapatite (HAP), 2-hydroxyethyl-methacrylate (HEMA), 2-isocyanatoethyl methacrylate (IEM), methanol (MeOH), pyridinium p-toluensulfonate, self-etch adhesive (SEA), shear bond strength (SBS), sodium sulfate (Na2SO4), tetrabutylammonium fluoride, tetrahydrofuran (THF), tetramethylsilane (TMS), triethylamine (TEA), trimethylsilyl bromide (TMSBr), and triethylene glycol dimethacrylate (TEGDMA).
2.2. Materials
Excepted for the preparation of compounds 1b, 2b, 8a, and 8b, all reactions were carried out under an argon atmosphere. DCM and MeOH were dried over molecular sieves. Dry THF was purchased from Sigma-Aldrich. Diethyl 2-aminoethylphosphonate 2a,[Citation20] diethyl 6-bromohexylphosphonate,[Citation21] 10-(methacryloyloxy)decylphosphonic acid 10,[Citation22], and N-(2-aminoethyl)methacrylamide 7 [Citation23] were prepared according to the literature. TMSBr (Sigma-Aldrich, Switzerland) was distilled prior to use. N-Succinimidyl-methacrylate and IEM were purchased from TCI. Irgacure 819 was purchased from Ciba. Bis-GMA (Evonic, Germany), Ivocerin® (Ivoclar Vivadent AG, Liechtenstein), DEBAAP (Ivoclar Vivadent AG, Liechtenstein), CQ (Rahn, Switzerland), EMBO (Sigma-Aldrich, Switzerland), and BHT (Sigma-Aldrich, Switzerland) were used in the adhesive formulations and for the photopolymerization studies. Thin layer chromatography was performed on silica gel 60 F-254 plates. Column chromatographies were performed on Roth silica gel 60 (30–200 μm). All other reagents used in the syntheses were purchased from Sigma-Aldrich (Switzerland) and were used without further purification.
2.3. Methods
1H NMR, 13C NMR, and 31P NMR spectra were recorded on a DPX-400 spectrometer using TMS as internal reference for 1H NMR and 13C NMR spectroscopic chemical shifts and using H3PO4 (85%) as external reference for 31P NMR spectroscopic chemical shifts. Data are given in the following order: chemical shift in ppm, multiplicity (s, singlet; ls, large singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet), coupling constant in Hertz, assignment. High-resolution mass spectra (HRMS) were obtained with a Waters Q-TOF Micro instrument in positive- or negative-ion mode electrospray ionization and lockspray with orthophosphoric acid. These analyses were performed with an infusion rate of 5 μL min−1, a source temperature of 80 °C, and a desolvation temperature of 120 °C.
2.4. Syntheses
2.4.1. Synthesis of acidic monomers 4a and 4b
Diethyl 6-phthalimidohexylphosphonate 1b
Diethyl 6-bromohexylphosphonate 5 (15.0 g, 49.8 mmol) was added to a solution of potassium phthalimide (13.8 g, 74.7 mmol, 1.5 eq.) in DMF (100 mL). The mixture was stirred for 17 h at 100 °C. The solution was filtered and concentrated under reduced pressure. EA (100 mL) was added to the crude product. The solution was filtered and washed with distilled water (2 × 100 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA). 13.14 g (35.8 mmol) of the desired product was isolated.
Yield: 72%. Aspect: slightly yellow liquid. 1H NMR (400 MHz, CDCl3): δ = 1.28–1.48 (m, 4H, CH2); 1.31 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 1.53–1.77 (m, 6H, CH2); 3.68 (t, 3JHH = 7.2 Hz, 2H, CH2N); 4.00–4.15 (m, 4H, POCH2CH3); 7.67–7.74 (m, 2H, CHAr); 7.81–7.88 (m, 2H, CHAr). 31P NMR (162 MHz, CDCl3): δ = 32.3. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCP = 6.0 Hz, POCH2CH3); 22.3 (d, 2JCP = 5.3 Hz, CH2CH2P); 25.6 (d, 1JCP = 140.6 Hz, CH2P); 26.3 (CH2); 28.3 (CH2); 30.1 (d, 3JCP = 17.1 Hz, CH2CH2CH2P); 37.8 (CH2N); 61.3 (d, 2JCP = 6.4 Hz, POCH2CH3); 123.1 (CHAr); 132.1 (CAr); 133.8 (CHAr); 168.4 (C=O).
Diethyl 6-aminohexylphosphonate 2b
Hydrazine monohydrate (2.66 g, 53.1 mmol, 1.5 eq.) was added to a solution of phosphonate 1b (13.0 g, 35.4 mmol) in EtOH (135 mL). The mixture was refluxed for 2 h and the solvent was removed under reduced pressure. A solution of NaOH (10 wt% in distilled water, 250 mL) was added and the mixture was extracted with DCM (3 × 200 mL). The combined organic layers were dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. 7.37 g (31.1 mmol) of the desired product was isolated.
Yield: 88%. Aspect: colorless liquid. 1H NMR (400 MHz, CDCl3): δ = 1.25–1.45 (m, 6H, CH2); 1.28 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 1.51–1.75 (m, 4H, CH2); 2.64 (t, 3JHH = 7.0 Hz, 2H, CH2N); 3.98–4.13 (m, 4H, POCH2CH3). 31P NMR (162 MHz, CDCl3): δ = 32.4. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCP = 5.9 Hz, POCH2CH3); 22.4 (d, 2JCP = 5.4 Hz, CH2CH2P); 25.6 (d, 1JCP = 140.6 Hz, CH2P); 26.4 (CH2); 30.4 (d, 3JCP = 16.8 Hz, CH2CH2CH2P); 33.5 (CH2); 42.1 (CH2N); 61.3 (d, 2JCP = 6.5 Hz, POCH2CH3).
Diethyl 2-[3-(2-(methacryloyloxy)ethyl)ureido]-ethylphosphonate 3a
General procedure A. IEM (4.7 mL, 33.3 mmol, 1.0 eq.) was added dropwise to a stirred solution of phosphonate 2a (6.03 g, 33.3 mmol) in DCM (70 mL) at 0 °C. After complete addition, the mixture was stirred for 15 min at 0 °C and for 2 h at room temperature. After removal of the solvent under reduced pressure, the product was purified by flash column chromatography (eluent: EA/MeOH: 90/10). 10.97 g (32.6 mmol) of the desired monomer was isolated.
Yield: 98%. Aspect: colorless liquid. 1H NMR (400 MHz, CDCl3): δ = 1.28 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 1.90 (s, 3H, CH3); 1.97 (dt, 3JHH = 6.6 Hz, 2JHP = 17.3 Hz, 2H, CH2P); 3.36–3.49 (m, 4H, CH2N); 3.96–4.10 (m, 4H, POCH2CH3); 4.16 (t, 3JHH = 5.6 Hz, 2H, OCH2CH2NH); 5.46 (t, 3JHH = 5.8 Hz, 1H, NH); 5.52–5.55 (m, 1H, C=CH2); 5.75 (t, 3JHH = 5.8 Hz, 1H, NH); 6.08 (s, 1H, C=CH2). 31P NMR (162 MHz, CDCl3): δ = 30.5. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCP = 6.2 Hz, POCH2CH3); 18.3 (CH3); 26.2 (d, 1JCP = 138.4 Hz, CH2P); 34.3 (d, 2JCP = 5.2 Hz, NHCH2CH2P); 39.2 (OCH2CH2NH); 61.8 (d, 2JCP = 6.5 Hz, POCH2CH3); 64.2 (OCH2CH2NH); 125.8 (C=CH2); 136.1 (C=CH2); 158.2 (NHC=O); 167.3 (C=O). HRMS (m/z): Calcd for C13H26N2O6P, 337.1529; Found, 337.1536 [M + H]+.
Diethyl 6-[3-(2-(methacryloyloxy)ethyl)ureido]-hexylphosphonate 3b
Methacrylate 3b was prepared from phosphonate 2b (17.29 g, 72.9 mmol) according to the general procedure A. 27.75 g (70.7 mmol) of the desired product was isolated.
Yield: 97%. Aspect: colorless oil. 1H NMR (400 MHz, CDCl3): δ = 1.24–1.49 (m, 6H, CH2); 1.29 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 1.50–1.62 (m, 2H, CH2); 1.63–1.75 (m, 2H, CH2); 1.91 (s, 3H, CH3); 3.11 (apparent as q, 3JHH = 6.8 Hz, 2H, CH2N); 3.45 (apparent as q, 3JHH = 5.6 Hz, 2H, CH2N); 3.96–4.10 (m, 4H, POCH2CH3); 4.17 (t, 3JHH = 5.6 Hz, 2H, OCH2CH2NH); 5.22 (t, 3JHH = 5.4 Hz, 1H, NH); 5.32 (t, 3JHH = 5.8 Hz, 1H, NH); 5.53–5.56 (m, 1H, C=CH2); 6.09 (s, 1H, C=CH2). 31P NMR (162 MHz, CDCl3): δ = 32.4. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCP = 6.1 Hz, POCH2CH3); 18.3 (CH3); 22.2 (d, 2JCP = 5.2 Hz, CH2CH2P); 25.2 (d, 1JCP = 140.4 Hz, CH2P); 26.2 (CH2); 29.7 (CH2); 29.9 (d, 3JCP = 15.8 Hz, CH2CH2CH2P); 39.2 (CH2N); 40.0 (CH2N); 61.5 (d, 2JCP = 6.6 Hz, POCH2CH3); 64.4 (OCH2CH2N); 125.8 (C=CH2); 136.1 (C=CH2); 158.5 (NHC=O); 167.4 (C=O). HRMS (m/z): Calcd for C17H34N2O6P, 393.2155; Found, 393.2151 [M + H]+.
2-[3-(2-(methacryloyloxy)ethyl)ureido]-ethylphosphonic acid 4a
General Procedure B. TMSBr (6.2 g, 44.7 mmol, 3.0 eq.) was added to a solution of phosphonate 3a (5.0 g, 14.9 mmol) in DCM (20 mL). The mixture was stirred for 5 h at 30 °C. The solvent and the excess of TMSBr were removed under reduced pressure. MeOH (20 mL) was added and the mixture was stirred for 15 min. The solution was concentrated under reduced pressure and the product was dried to a constant weight under high vacuum (ca. 0.1 mbar). 4.18 g (14.9 mmol) of the desired product was isolated.
Yield: 100%. Aspect: yellow high viscous oil. 1H NMR (400 MHz, MeOD): δ = 1.93 (s, 3H, CH3); 1.96–2.07 (m, 2H, CH2P); 3.40–3.50 (m, 4H, CH2N); 4.21 (t, 3JHH = 5.4 Hz, 2H, OCH2CH2NH); 5.63–5.66 (m, 1H, C=CH2); 6.12–6.15 (m, 1H, C=CH2). 31P NMR (162 MHz, MeOD): δ = 27.1. 13C NMR (101 MHz, MeOD): δ = 18.4 (CH3); 28.9 (d, 1JCP = 135.8 Hz, CH2P); 36.1 (NHCH2CH2P); 40.5 (OCH2CH2NH); 64.7 (OCH2CH2NH); 126.5 (C=CH2); 137.5 (C=CH2); 160.8 (NHC=O); 168.6 (C=O). HRMS (m/z): Calcd for C9H16N2O6P, 279.0746; Found, 279.0743 [M − H]−.
6-[3-(2-(methacryloyloxy)ethyl)ureido]-hexylphosphonic acid 4b
Phosphonic acid 4b was prepared from phosphonate 3b (5.9 g, 15.0 mmol) according to the general procedure B. 5.03 g (14.9 mmol) of the desired product was isolated.
Yield: 99%. Aspect: yellow high viscous oil. 1H NMR (400 MHz, MeOD): δ = 1.32–1.68 (m, 8H, CH2); 1.69–1.81 (m, 2H, CH2); 1.94 (s, 3H, CH3); 3.20 (t, 3JHH = 7.1 Hz, 2H, CH2N); 3.52 (t, 3JHH = 5.4 Hz, 2H, CH2N); 4.24 (t, 3JHH = 5.4 Hz, 2H, OCH2CH2NH); 5.64–5.67 (m, 1H, C=CH2); 6.13 (s, 1H, C=CH2). 31P NMR (162 MHz, MeOD): δ = 31.6. 13C NMR (101 MHz, MeOD): δ = 17.1 (CH3); 22.2 (d, 2JCP = 4.8 Hz, CH2CH2P); 25.9 (CH2); 26.2 (d, 1JCP = 137.7 Hz, CH2P); 29.0 (CH2); 29.8 (d, 3JCP = 16.7 Hz, CH2CH2CH2P); 39.4 (CH2N); 40.4 (CH2N); 63.0 (OCH2CH2N); 125.3 (C=CH2); 136.1 (C=CH2); 159.5 (NHC=O); 167.2 (C=O). HRMS (m/z): Calcd for C13H24N2O6P, 335.1372; Found, 335.1373 [M − H]−.
2.4.2. Synthesis of acidic monomers 9a and 9b
Diethyl 2-[(1H-imidazole-1-yl-carbonyl)amino]-ethylphosphonate 6a
General Procedure C. A solution of diethyl 2-aminoethylphosphonate 2a (7.0 g, 38.6 mmol) in dry THF (190 mL) was cooled down to −10 °C. A −10 °C cold solution of CDI (7.5 g, 46.4 mmol, 1.2 eq.) in dry THF (270 mL) was added slowly over 20 min. The mixture was stirred for 1.5 h (the temperature staid between −10 °C and 0 °C). The reaction mixture was then allowed to warm up to room temperature and was stirred for 15 h. Distilled water (5 mL) was added in order to quench the excess of CDI. The solvent was removed under reduced pressure to give 15.1 g of a slightly yellow oil. The crude product was used in the next step without further purification.
Aspect: slightly yellow oil. 1H NMR (400 MHz, CDCl3): δ = 1.35 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 2.08 (dt, 3JHH = 6.4 Hz, 2JHP = 17.2 Hz, 2H, CH2P); 3.65–3.80 (m, 2H, NCH2CH2P); 4.06–4.22 (m, 4H, POCH2CH3); 7.07–7.09 (m, 1H, CHN); 7.38–7.39 (m, 1H, CHN); 7.48 (ls, 1H, NH) 8.16–8.18 (m, 1H, CHN). 31P NMR (162 MHz, CDCl3): δ = 29.9. 13C NMR (101 MHz, MeOD): δ = 16.4 (d, 3JCP = 5.8 Hz, POCH2CH3); 25.0 (d, 1JCP = 138.3 Hz, CH2P); 34.8 (d, 2JCP = 6.4 Hz, CH2CH2P); 62.3 (d, 2JCP = 7.2 Hz, POCH2CH3); 115.8 (CHN); 130.6 (CHN); 136.1 (CHN); 148.8 (CO).
Diethyl 6-[(1H-imidazole-1-yl-carbonyl)amino]-hexylphosphonate 6b
Phosphonate 6b was prepared from phosphonate 2b (4.75 g, 20.0 mmol) according to the general procedure C. 8.27 g of crude product was isolated.
Aspect: slightly yellow oil. 1H NMR (400 MHz, MeOD): δ = 1.31 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 1.36–1.52 (m, 4H, CH2); 1.53–1.68 (m, 4H, CH2); 1.73–1.85 (m, 2H, CH2); 3.35 (t, 3JHH = 7.1 Hz, 2H, NCH2); 4.00–4.15 (m, 4H, POCH2CH3); 7.04–7.06 (m, 1H, CHN); 7.60–7.62 (m, 1H, CHN); 8.23–8.25 (m, 1H, CHN). 31P NMR (162 MHz, CDCl3): δ = 33.5. 13C NMR (101 MHz, CDCl3): δ = 15.3 (d, 3JCP = 6.1 Hz, POCH2CH3); 21.9 (d, 3JCP = 6.1 Hz, CH2CH2CH2P); 24.2 (d, 1JCP = 140.7 Hz, CH2P); 25.9 (CH2); 28.7 (CH2); 29.6 (d, 2JCP = 15.9 Hz, CH2CH2P); 40.3 (CH2N); 61.7 (d, 2JCP = 6.4 Hz, POCH2CH3); 116.4 (CHN); 128.7 (CHN); 136.0 (CHN); 149.3 (CO).
Diethyl 2-[3-(2-methacrylamidoethyl)ureido]-ethylphosphonate 8a
General procedure D. N-(2-aminoethyl)methacrylamide 7 (1.8 g, 14.4 mmol) was added to a solution of crude phosphonate 6a (5.0 g) in THF (40 mL) and the solution was stirred for 15 h at room temperature. The solvent was removed under reduced pressure and the crude product was purified by flash column chromatography (eluent: hexane/MeOH/DCM: 5/1/4). 3.04 g (9.1 mmol) of the desired monomer was isolated.
Yield: 73% (from 2a over 2 steps). Aspect: slightly yellow high viscous oil. 1H NMR (400 MHz, MeOD): δ = 1.32 (t, 3JHH = 7.0 Hz, 6H, POCH2CH3); 1.92–1.95 (m, 3H, CH3); 2.03 (dt, 3JHH = 7.4 Hz, 2JHP = 20.2 Hz, 2H, CH2P); 3.23–3.41 (m, 6H, CH2N); 4.04–4.16 (m, 4H, POCH2CH3); 5.36–5.39 (m, 1H, C=CH2); 5.71–5.74 (s, 1H, C=CH2). 31P NMR (162 MHz, CDCl3): δ = 30.2. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCP = 6.1 Hz, POCH2CH3); 18.6 (CH3); 26.3 (d, 1JCP = 138.4 Hz, CH2P); 34.4 (d, 2JCP = 4.8 Hz, CH2CH2P); 39.3 (CH2N); 42.1 (CH2N); 61.9 (d, 2JCP = 6.4 Hz, POCH2CH3); 120.0 (C=CH2); 139.5 (C=CH2); 159.6 (CCON); 168.9 (NCON). HRMS (m/z): Calcd for C13H27N3O5P, 336.1688; Found, 336.1689 [M + H]+.
Diethyl 6-[3-(2-methacrylamidoethyl)ureido]-hexylphosphonate 8b
Phosphonate 8b was prepared from crude phosphonate 6b (8.27 g) and N-(2-aminoethyl)methacrylamide 7 (2.74 g, 21.4 mmol) according to the general procedure D. 5.76 g (14.7 mmol) of the desired monomer was isolated.
Aspect: slightly yellow viscous oil. Yield: 79% (from 2b over 2 steps). Aspect: slightly yellow viscous oil. 1H NMR (400 MHz, CDCl3): δ = 1.33 (t, 3JHH = 7.1 Hz, 6H, POCH2CH3); 1.28–1.54 (m, 6H, CH2); 1.54–1.68 (m, 2H, CH2); 1.68–1.80 (m, 2H, CH2); 1.96 (s, 3H, CH3); 3.16 (q, 3JHH = 6.4 Hz, 2H, NCH2CH2CH2); 3.34–3.40 (m, 4H, NCH2CH2N); 4.00–4.15 (m, 4H, POCH2CH3); 5.04 (ls, 1H, NH); 5.31–5.34 (m, 1H, C=CH2); 5.37 (ls, 1H, NH); 5.77–5.79 (m, 1H, C=CH2); 7.29 (ls, 1H, NH). 31P NMR (162 MHz, CDCl3): δ = 32.5. 13C NMR (101 MHz, CDCl3): δ = 16.5 (d, 3JCP = 6.0 Hz, POCH2CH3); 18.6 (CH3); 22.2 (d, 3JCP = 5.0 Hz, PCH2CH2CH2); 25.2 (d, 1JCP = 140.4 Hz, CH2P); 26.2 (CH2); 29.7 (CH2); 29.9 (d, 2JCP = 15.4 Hz, CH2CH2P); 39.3 (CH2N); 40.7 (CH2N); 42.7 (CH2N); 61.6 (d, 2JCP = 6.6 Hz, POCH2CH3); 120.1 (C=CH2); 139.4 (CH2=C); 160.0 (CCON); 169.1 (NCON). HRMS (m/z): Calcd for C17H35N3O5P, 392.2314; Found, 392.2312 [M + H]+.
2-[3-(2-methacrylamidoethyl)ureido]-ethylphosphonic acid 9a
Phosphonic acid 9a was prepared from phosphonate 8a (360 mg, 1.07 mmol) according to general procedure B. 300 mg (1.07 mmol) of the desired product was isolated.
Yield: quantitative. Aspect: hygroscopic white powder. 1H NMR (400 MHz, D2O): δ = 1.84 (s, 3H, CH3); 1.92 (dt, 3JHH = 7.3 Hz, 2JHP = 18.1 Hz, 2H, CH2P); 3.17–3.33 (m, 6H, CH2N); 5.37 (s, 1H, C=CH2); 5.60 (s, 1H, C=CH2). 31P NMR (162 MHz, D2O): δ = 27.8. 13C NMR (101 MHz, D2O): δ = 17.6 (CH3); 27.4 (d, 1JCP = 132.9 Hz, CH2P); 34.3 (d, 2JCP = 2.3 Hz, CH2CH2P); 38.9 (CH2N); 39.6 (CH2N); 121.00 (C=CH2); 138.9 (CH2=C); 160.1 (CCON); 172.0 (NCON). HRMS (m/z): Calcd for C9H17N3O5P, 278.0906; Found, 278.0.904 [M − H]−.
6-[3-(2-methacrylamidoethyl)ureido]-hexylphosphonic acid 9b
Phosphonic acid 9b was prepared from phosphonate 8b (2.6 g, 6.64 mmol) according to the general procedure B. 2.23 g (6.64 mmol) of the desired product was isolated.
Yield: quantitative. Aspect: slightly yellow hygroscopic solid. 1H NMR (400 MHz, D2O): δ = 1.19–1.58 (m, 8H, CH2); 1.65–1.80 (m, 2H, CH2); 1.86 (s, 3H, CH2=CCH3); 3.01 (t, 3JHH = 6.9 Hz, 2H, NCH2CH2CH2); 3.20–3.34 (m, 4H, NCH2CH2N); 5.39 (s, 1H, C=CH2); 5.63 (s, 1H, C=CH2). 31P NMR (162 MHz, D2O): δ = 32.5. 13C NMR (101 MHz, D2O): δ = 17.6 (CH3); 22.8 (d, 3JCP = 4.9 Hz, CH2CH2CH2P); 25.3 (CH2); 25.9 (d, 1JCP = 133.7 Hz, CH2P); 28.8 (CH2); 29.3 (d, 2JCP = 16.7 Hz, CH2CH2P); 38.9 (CH2N); 39.7 and 39.8 (CH2N); 121.0 (C=CH2); 138.9 (CH2=C); 160.5 (CCON); 172.0 (NCON). HRMS (m/z): Calcd for C13H25N3O5P, 334.1532; Found, 334.1533 [M − H]−.
2.4.3. Synthesis of BEA
A solution of N-butyl-N-ethylamine (20.0 g, 198 mmol, 1.05 eq.) and TEA (20.0 g, 198 mmol, 1.05 eq.) in dry DCM (400 mL) was cooled to 0 °C. Acryloyl chloride (17.0 g, 188 mmol) in dry DCM (200 mL) was added dropwise over 1.5 h. The mixture was stirred for 30 min at 0 °C and was then allowed to reach room temperature. After stirring for additional 2.5 h, distilled water (200 mL) was added. The two layers were separated and the organic layer was concentrated under reduced pressure. The 200-mL aqueous solution was added to the crude product. The solution was extracted with EA (3 × 200 mL). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by distillation under reduced pressure (T = 50 °C; p = 0.035 mbar). 24.8 g (160 mmol) of the desired product was isolated.
Yield: 85%. Aspect: colorless liquid. 1H NMR (400 MHz, CDCl3): δ = 0.93, 0.95 (2t, 3JHH = 6.9 Hz, 3H, CH2CH2CH3); 1.16, 1.19 (2t, 3JHH = 7.5 Hz, 3H, NCH2CH3); 1.28–1.39 (m, 2H, CH2); 1.50–1.62 (m, 2H, CH2); 3.30, 3.38 (2t, 3JHH = 7.9 Hz, 2H, NCH2CH2); 3.39, 3.44 (2q, 3JHH = 7.2 Hz, 2H, NCH2CH3); 5.66 (dm, 3JHH = 10.5 Hz, 1H, CH=CH2); 6.35 (dm, 3JHH = 16.7 Hz, 1H, CH=CH2); 6.54, 6.56 (2dd, 3JHH = 16.7 Hz, 3JHH = 10.5 Hz, 1H, CH=CH2). 13C NMR (101 MHz, CDCl3): δ = 12.9, 14.7 (CH3); 13.8, 13.9 (CH3); 20.0, 20.2 (CH2); 30.0, 31.8 (CH2); 41.2, 45.9 (CH2N) 42.5, 47.4 (CH2N); 127.1, 127.5 (CH=CH2); 127.8, 127.9 (CH=CH2); 165.7, 165.7 (CO).
2.5. Photopolymerization procedure
Photopolymerizations were carried out with a Perkin Elmer differential scanning calorimeter (DSC, Pyris Diamond). 0.2 mol% Ivocerin® was added to each mixture as a photoinitiator. The experiments were performed under a stream of nitrogen. A sample (ca. 1.0 mg) of each mixture was placed in an uncovered aluminum DSC pan. The DSC chamber was purged with nitrogen for 4 min. The acquisition was then started. After 1 min of acquisition, the samples were irradiated for 2 min at 37 °C with a LED curing light (Bluephase, monowave LED with a maximum intensity at 470 nm, Ivoclar Vivadent AG). The incident light intensity was about 20 mW cm−2. Each experiment was repeated three times. The heat flux was monitored as a function of time using the DSC under isothermal conditions. Double-bond conversion (DBC) was calculated as the quotient of the overall enthalpy evolved [ΔHp (J g−1)] and the theoretical enthalpy obtained for 100% conversion of the mixtures [ΔH0p (J g−1)] (Equation (1)):(1)
ΔH0p was calculated according to the following formula (Equation (2)):(2)
where ΔH0i is the theoretical enthalpy of monomer i (i = monomethacrylate and monomethacrylamide, ΔH0i = 54.8 kJ mol−1,[Citation24] i = dimethacrylate, ΔH0i = 109.7 kJ mol−1; i = BEA, ΔH0i = 74.7 kJ mol−1,[Citation13] Mi its molar mass and Pi is the amount used in the formulation (wt%). The rate of polymerization (Rp) was calculated according to the following formula (Equation (3)):(3)
where Q is the heat flow per s during the reaction and m is the mass of the mixture in the sample.
2.6. SBS measurement [Citation25]
Freshly extracted bovine mandibular incisors were embedded in unsaturated polyester resin (ViscoVoss). Flat dentinal and enamel surfaces were prepared with 120-grit and 400-grit wet silicon carbide paper on the labial side of the embedded teeth. The adhesive was first rubbed on the prepared surface (dentin or enamel) with a microbrush for 20 s. The adhesive layer was strongly air-dried and light-cured for 10 s with a LED curing light (Bluephase G2, polywave LED with a broadband spectrum from 385 to 515 nm and 2 maxima at 410 and 470 nm, Ivoclar Vivadent AG). A polyethylene mold with a central 2.38-mm diameter circular hole was fixed on the surface. A composite (Tetric EvoCeram®, Ivoclar Vivadent AG) was inserted in the mold and light-cured for 20 s. The samples were subsequently stored in water at 37 °C for 24 h before being tested. The SBS was measured using a universal testing machine (Zwick, Germany) at a crosshead speed of 0.8 mm min−1. Ten samples were tested for each adhesive.
3. Results and discussion
3.1. Syntheses
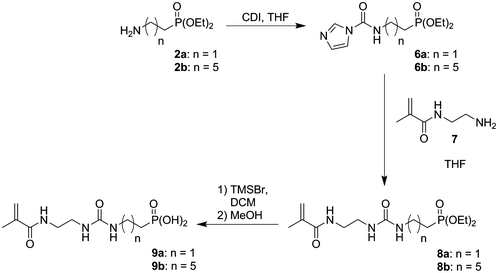
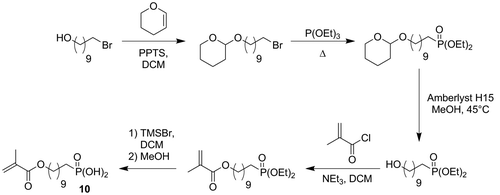
Acidic monomers 4a and 4b were synthesized in 4 and 5 steps, respectively (Scheme ). First of all, amine 2a and bromophosphate 5 were prepared according to literature procedures.[Citation20,21] Aminophosphonate 2b was then obtained from phosphonate 5 by a Gabriel synthesis. Bromophosphonate 5 was treated with potassium phthalimide in DMF at 100 °C to provide phthalimide 1b in 72% yield. Cleavage of the phtalimide group by hydrazine led to the desired amine 2b in 88% yield. Both amines 2a and 2b were subsequently reacted with IEM to give the desired monomers 3a and 3b in excellent yields (>97%). Silylation of 3a and 3b using TMSBr, followed by the methanolysis of the silyl ether, finally provided acidic monomers 4a and 4b. Phosphonic acids 4a and 4b were isolated in 63% and 41% overall yields, respectively. It is well known that (meth)acrylates are not hydrolytically stable during the storage of some SEAs.[Citation1,26,27] Indeed, SEAs are acidic aqueous solutions. In these conditions of storage, (meth)acrylates might undergo hydrolysis, leading to a modification of the chemical composition of the adhesive. In order to circumvent this problem, hydrolytically stable monomers have been incorporated in SEAs. Among these monomers, (meth)acrylamides were the most efficient.[Citation11,21,28–31] The second part of our synthetic work consisted therefore in the preparation of methacrylamides 9a and 9b. Those monomers were synthesized in three steps starting from amines 2a and 2b (Scheme ). N-substituted carbonylimidazolides 6a and 6b were obtained by reacting amines 2a and 2b with CDI in THF. The addition of N-(2-aminoethyl)methacrylamide 7 to crude phosphonates 6a and 6b in THF led to the formation of urea monomers 8a and 8b. Deprotection of the phosphonate groups finally provided the expected acidic monomers 9a and 9b. Phosphonic acids 9a and 9b were isolated in 47% and 34% overall yields, respectively. In order to study the influence of the urea group on both the reactivity and the adhesive properties of the new monomers, phosphonic acid 10 was also synthesized (Scheme ). This monomer, which spacer is a decyl group, was prepared according to the literature procedure.[Citation22]
Scheme 1. Synthesis of monomers 4a and 4b.
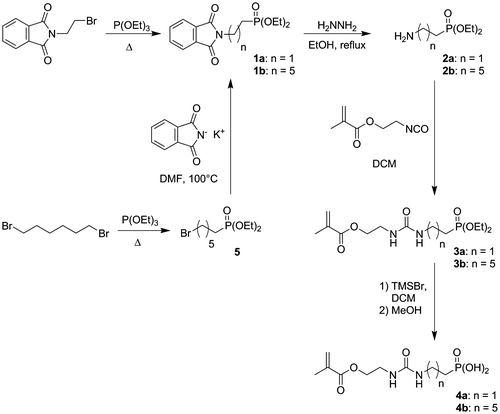
Scheme 2. Synthesis of monomers 9a and 9b.
Scheme 3. Synthesis of monomer 10.
All new monomers were characterized by 1H NMR, 31P NMR, and 13C NMR spectroscopy. For example, the 1H NMR spectrum of monomer 4b is shown in Figure . It is characterized by the alkyl spacer protons at 1.32–1.81 ppm (10H, m), the methylene groups in alpha position of the urea moiety at 3.20 and 3.52 ppm (4H, t), and the protons next to the ester group at 4.24 ppm (2H, t). The protons of the methacrylate group are identified by the presence of signals at 1.94 (methyl group), 5.64–5.67, and 6.13 ppm. Monomers 4a, 4b, 9a, and 9b are all soluble in a mixture of water and ethanol.
3.2. Hydrolytic stability
In order to investigate the hydrolytic stability of acidic monomers 4a, 4b, 9a, 9b, and 10, 20 wt% solutions in D2O/EtOD6 (1/1: w/w) were prepared. The solutions were stored at 37 °C for 15 d and the hydrolysis was monitored by 1H NMR spectroscopy. Results are shown in Figure . Methacrylate monomers containing a urea group (4a and 4b) were highly instable and almost completely hydrolyzed after 15 d. On the other hand, methacrylate 10 (alkyl spacer group) was shown to be significantly more stable than monomers 4a and 4b. Therefore, the presence of the urea group leads to an acceleration of the hydrolysis. This phenomenon is most likely due to hydrogen bonding interactions between the urea moiety and the carbonyl of the methacrylate group. As expected, methacrylamides 9a and 9b were stable in aqueous solutions. Even after 2 months of storage, only 3% of each monomer underwent hydrolysis. These results clearly demonstrate that acidic monomers 9a and 9b are better candidates than methacrylates 4a and 4b to be incorporated in SEA formulations.
3.3. Photopolymerization in bulk
Photopolymerization investigations were carried out in order to assess the reactivity of the synthesized monomers. Each monomer or monomer mixture was polymerized under the same conditions (irradiation time: 2 min; light intensity: 20 mW cm−2). Ivocerin® (0.2 mol%) was added as photoinitiator.[Citation32] Homopolymerization of phosphonates 3a, 3b, 8a, and 8b was first studied. As acidic monomers 4a, 4b, 9a, and 9b are either highly viscous oils or solids, their homopolymerization could not be investigated. HEMA and TEGDMA, monomers which are often used in dental materials, were also homopolymerized. Figures and , respectively, show the DBC and the rate of polymerization (Rp) for each monomer as a function of time. Urea monomers 3a, 3b, 8a, and 8b were significantly more reactive than HEMA. They also polymerized faster than TEGDMA, a crosslinking dimethacrylate. They behave consequently like multifunctional monomers. Avci et al. observed a similar phenomenon as they studied the homopolymerization of some urea methacrylates.[Citation14] The ability of the urea group to form hydrogen bonds has been shown to be partially responsible for this high reactivity. This is mainly due to a preorganization of the monomers (increase in the propagation reaction rate constant) as well as to a restriction of the mobility. Abstraction of the labile protons in the alpha position of the urea moiety (chain transfer reaction) could also result in an acceleration of the polymerization. Among the urea monomers, methacrylates 3a ( = 5.2 s, Rpmax = 0.096 s−1) and 3b (
= 4.9 s, Rpmax = 0.081 s−1) were the most reactive. After 2 min of irradiation, DBCs measured for methacrylate monomers 3a (93.6%) and 3b (94.8%) were significantly higher than for methacrylamides 8a (71.1%) and 8b (77.7%). This is probably caused by the hydrogen bonding capacity of the methacrylamide group, leading to an additional decrease in the monomer mobility. Interestingly, the final DBCs obtained with urea methacrylates 3a and 3b were also much higher than for TEGDMA (68.9%). Therefore, these highly reactive monomethacrylates may be used to replace dimethacrylate (e.g. TEGDMA) diluents in dental materials.
Methacrylates 3a, 3b, 4a, 4b, and 10 were then copolymerized with BEA (2/8: mol/mol) (Figures and ). The results showed that phosphonic acids 4a and 4b are significantly more reactive than their corresponding phosphonates 3a and 3b. Moreover, the final DBCs measured for the mixtures containing acidic monomers 4a and 4b were the lowest (67.1 and 71.3%). Once again, these observations can be explained by the ability of the phosphonic acid group to form additional hydrogen bonds. In order to study more precisely the influence of the urea group on the monomer reactivity, phosphonic acid 10 (alkyl spacer group) was also copolymerized with BEA. The mixture containing urea methacrylate 4b ( = 4.7 s, Rpmax = 0.095 s−1) was found to be significantly more reactive than the one based on phosphonic acid 10 (
= 15.2 s, Rpmax = 0.059 s−1). This result confirms that the presence of a urea group leads to an increase in the reactivity. Copolymerization of BEA with 3b provided unexpected results (Figure ). Indeed, the polymerization of the mixture BEA/3b (8/2) (
= 17.1 s, Rpmax = 0.057 s−1) was significantly slower than the homopolymerization of either monomer 3b (
= 4.9 s, Rpmax = 0.081 s−1) or BEA (
= 9.1 s, Rpmax = 0.087 s−1).
Copolymerizations with various proportions of BEA and 3b (BEA/3b: 8/2, 1/1 and 2/8) were subsequently carried out (Figures and ). All copolymerizations led to nearly full conversion. The results showed that the higher the amount of urea monomer 3b, the faster the polymerization. As already stated, the high polymerization rate of urea methacrylate 3b might be due to a preorganization of the monomer thanks to the formation of hydrogen bonds. The addition of BEA to 3b might disturb this preorganization and therefore result in a decrease in the polymerization rate. BEA polymerized faster than the mixtures BEA/3b (8/2) and BEA/3b (1/1). The higher the proportions of BEA in the mixture, the weaker the effect of hydrogen bonding on the polymerization rate. In this case, the reactivity of methacrylate 3b is significantly reduced. Copolymerization of methacrylamides 8a, 8b, 9a, and 9b with BEA was also investigated (Figures and ). As already observed for the synthesized methacrylates (Figures and ), phosphonic acids 9a and 9b were more reactive than their corresponding phosphonates 8a and 8b. It can also be noticed that mixtures BEA/8a ( = 5.5 s, Rpmax = 0.087 s−1) and BEA/8b (
= 5.4 s, Rpmax = 0.089 s−1) polymerized significantly faster than BEA/3a (
= 17.6 s, Rpmax = 0.052 s−1) and BEA/3b (
= 17.1 s, Rpmax = 0.057 s−1). The higher reactivity of methacrylamides 8a and 8b in copolymerization with BEA is probably due to the hydrogen bonding capacity of the methacrylamide group.
3.4. Adhesive properties
Five SEA formulations were prepared in order to investigate the adhesive properties of acidic monomers 4a, 4b, 9a, 9b, and 10 (Table ). Each SEA contained an acidic monomer, crosslinking monomers (Bis-GMA and DEBAAP), photoinitiators (CQ/EMBO and Irgacure 819), solvents (water and i-PrOH), and additives (Aerosil and BHT as inhibitor). These adhesives were used to mediate a bond between a composite (Tetric EvoCeram®) and dental hard tissues (dentin and enamel). SBSs were measured in order to evaluate the adherence (Table ). Each adhesive was able to achieve a strong bond between dentin/enamel and the restorative material. The nature of the polymerizable group does not seem to have any influence on the adhesion to dentin. Indeed, similar dentin SBS were obtained with SEAs 1 and 3. The same observation can be made with SEAs 2 and 4. However, the formulation based on methacrylate 4a (SEA 1) provided significantly higher enamel SBS than the one based on the corresponding methacrylamide 9a (SEA 3). The results also show that the longer the spacer group of the acidic monomer, the higher the enamel and dentin SBS. Similar trends have already been reported in the literature.[Citation33] This phenomenon was explained on the basis of the ‘adhesion-decalcification concept’. The increase in the spacer group length results in a decrease in the solubility of the acidic monomer calcium salt in the aqueous environment. As a consequence, the chemical adhesion of the acidic monomer to HAP is improved. SEA 2 (monomer 4b) led to higher dentin and enamel SBS than SEA 5 (monomer 10, alkyl spacer). The increase in the enamel SBS was particularly significant. This result highlights the strong influence of the urea group on the adhesion. The presence of the urea moiety probably improves, due to its metal chelating properties, the interaction with HAP. The ability of urea monomers to form hydrogen bonds may also enhance the mechanical properties of the adhesive.
Table 1. Composition of five experimental SEAs.
Table 2. Dentin and enamel shear bond strength tests.
4. Conclusion
Urea monomers bearing a phosphonic acid group 4a, 4b, 9a, and 9b as well as the 10-(methacryloyloxy)decylphosphonic acid 10 were successfully synthesized. Although methacrylates 4a and 4b underwent fast hydrolysis when stored in D2O/EtOD6, methacrylamides 9a and 9b were hydrolytically stable. The photopolymerization behavior of these acidic monomers as well as of their corresponding phosphonates 3a, 3b, 8a, and 8b were investigated. Urea monomers 3a, 3b, 8a, and 8b were more reactive than HEMA and TEGDMA, a crosslinking monomer. Homopolymerization of methacrylates 3a and 3b led to almost complete conversion, whereas final DBCs of 71.1% and 77.7% were obtained for the corresponding methacrylamides 8a and 8b, respectively. Copolymerization of the synthesized monomers with BEA showed that the acidic monomers were significantly more reactive than their corresponding phosphonates. The presence of a urea group was found to significantly enhance the rate of polymerization. This phenomenon was attributed to the hydrogen bonding capacity of the urea group as well as to potential chain transfer reactions. The adhesive properties of each acidic monomer were also evaluated. SEAs based on urea monomers 4a, 4b, 9a, and 9b were able to mediate a strong bond between a composite restorative material and dentin/enamel. It was shown that the presence of the urea moiety leads to a significant increase in both dentin and enamel SBS. The influence of the spacer group length on the adhesion was also studied. Results showed that monomers bearing longer spacer groups provide higher SBS. Due to its good hydrolytic stability, high reactivity, and excellent adhesive properties, methacrylamide 9b is a great candidate for the formulation of new SEAs.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Moszner N, Hirt T. New polymer-chemical developments in clinical dental polymer materials: enamel-dentin adhesives and restorative composites. J. Polym. Sci. A: Polym. Chem. 2012;50:4369–4402.10.1002/pola.v50.21
- Van Meerbeek B, Yoshihara K, Yoshida Y, et al. State of the art of self-etch adhesives. Dent. Mater. 2011;27:17–28.10.1016/j.dental.2010.10.023
- Van Landuyt KL, Snauwaert J, De Munck J, et al. Systematic review of the chemical composition of contemporary dental adhesives. Biomaterials. 2007;28:3757–3785.10.1016/j.biomaterials.2007.04.044
- Moszner N, Catel Y. Phosphorus-based polymers: from synthesis to applications. In: Monge S, David G, editors, Cambridge: RSC polymer chemistry series. Vol. 11; 2014; Chapter 8. p.151–166.10.1039/2044-0804
- Moszner N, Salz U. Recent developments of new components for dental adhesives and composites. Macromol. Mater. Eng. 2007;292:245–271.10.1002/(ISSN)1439-2054
- Van Landuyt KL, Yoshida Y, Hirata I, et al. Influence of the chemical structure of functional monomers on their adhesive performance. J. Dent. Res.. 2008;87:757–761.10.1177/154405910808700804
- Yoshida Y, Van Meerbeek B, Nakayama Y, et al. Adhesion to and decalcification of hydroxyapatite by carboxylic acids. J. Dent. Res. 2001;80:1565–1569.10.1177/00220345010800061701
- Yoshida Y, Nagakane K, Fukuda R, et al. Comparative study on adhesive performance of functional monomers. J. Dent. Res. 2004;83:454–458.10.1177/154405910408300604
- Yoshihara K, Yoshida Y, Hayakawa S, et al. Self-etch monomer-calcium salt deposition on dentin. J. Dent. Res. 2011;90:602–606.10.1177/0022034510397197
- Bilgici ZS, Turker SB, Avci D. Novel bisphosphonated methacrylates: synthesis, polymerizations, and interactions with hydroxyapatite. Macromol. Chem. Phys. 2013;214:2324–2335.10.1002/macp.v214.20
- Catel Y, Degrange M, Le Pluart L, et al. Synthesis, photopolymerization, and adhesive properties of new bisphosphonic acid monomers for dental application. J. Polym. Sci. A: Polym. Chem. 2009;47:5258–5271.10.1002/pola.v47:20
- Catel Y, Fischer UK, Moszner N. Monomers for adhesive polymers, 13. 1 Synthesis, radical photopolymerization and adhesive properties of polymerizable 2-substituted 1,3-propylidenediphosphonic acids. Des. Monomers Polym. 2014;17:286–299.10.1080/15685551.2013.840503
- Catel Y, Bock T, Moszner N. Monomers for adhesive polymers, 15: synthesis, photopolymerization, and adhesive properties of polymerizable β-ketophosphonic acids. Polym. Chem J. Polym. Sci. A. 2014;52:3550–3563.
- Altin A, Akgun B, Buyukgumus O, et al. Synthesis and photopolymerization of novel, highly reactive phosphonated-urea-methacrylates for dental materials. React. Funct. Polym. 2013;73:1319–1326.10.1016/j.reactfunctpolym.2013.07.006
- Berchtold KA, Nie J, Stansbury JW, et al. Novel monovinyl methacrylic monomers containing secondary functionality for ultrarapid polymerization: steady-state evaluation. Macromolecules. 2004;37:3165–3179.10.1021/ma035862+
- Jansen JFGA, Dias AA, Dorschu M, et al. Fast monomers: factors affecting the inherent reactivity of acrylate monomers in photoinitiated acrylate polymerization. Macromolecules. 2003;36:3861–3873.10.1021/ma021785r
- Lee TY, Roper TM, Jönsson ES, et al. Influence of hydrogen bonding on photopolymerization rate of hydroxyalkyl acrylates. Macromolecules. 2004;37:3659–3665.10.1021/ma0305277
- Zhou H, Li Q, Lee TY, et al. Photopolymerization of acid containing monomers: real-time monitoring of polymerization rates. Macromolecules. 2006;39:8269–8273.10.1021/ma061332c
- Ibrahim OB, Refat MS, Salman M, et al. Chemical studies on the uses of urea complexes to synthesize compounds having electrical and biological applications. Int. J. Mater. Sci. 2012;2:67–82.
- Graillot A, Monge S, Faur C, et al. Synthesis by RAFT of innovative well-defined (co)polymers from a novel phosphorus-based acrylamide monomer. Polym. Chem. 2013;4:795–803.10.1039/C2PY20720F
- Catel Y, Degrange M, Le Pluart L, et al. Synthesis, photopolymerization and adhesive properties of new hydrolytically stable phosphonic acids for dental applications.. J. Polym. Sci. A: Polym. Chem. 2008;46:7074–7090.10.1002/pola.v46:21
- Derbanne MA, Besse V, Le Goff S, et al. Hydrolytically stable acidic monomers used in two steps self-etch adhesives. Polym. Degrad. Stab. 2013;98:1688–1698.10.1016/j.polymdegradstab.2013.06.006
- Tarasow TM, Tarasow SL, Tu C, et al. Characteristics of an RNA diels−alderase active site. J. Am. Chem. Soc. 1999;121:3614–3617.10.1021/ja983989m
- Anseth KS, Wang CM, Bowman CN. Kinetic evidence of reaction-diffusion during the polymerization of multi(meth)acrylate monomers. Macromolecules. 1994;27:650–655.10.1021/ma00081a004
- ISO29022: 2013.
- Salz U, Zimmermann J, Zeuner F, et al. Hydrolytic stability of self-etching adhesive systems. J. Adhes. Dent. 2005;7:107–116.
- Teshima I. J. Dent. Res. 2010;89:1281–1286.10.1177/0022034510379018
- Moszner N, Angermann J, Fischer U, et al. Monomers for adhesive polymers, 9-synthesis, radical photopolymerization, and properties of (meth)acrylamido dihydrogen phosphates. Macromol. Mater. Eng. 2013; 298: 454–461.10.1002/mame.v298.4
- Klee JE, Lehmann U. N-alkyl-N-(phosphonoethyl) substituted (meth)acrylamides – new adhesive monomers for self-etching self-priming one part dental adhesive. Beil. J. Org. Chem. 2009;5.
- Catel Y, Fischer UK, Moszner N. Monomers for adhesive polymers: 11. Structure–adhesive properties relationships of new hydrolytically stable acidic monomers. Polym. Int. 2013;62:1717–1728.10.1002/pi.2013.62.issue-12
- Altin A, Akgun B, Bilgici ZS, et al. Synthesis, photopolymerization, and adhesive properties of hydrolytically stable phosphonic acid-containing (meth)acrylamides. J. Polym. Sci. A: Polym. Chem. 2014;52:511–522.10.1002/pola.27025
- Moszner N, Zeuner F, Lamparth I, et al. Benzoylgermanium derivatives as novel visible-light photoinitiators for dental composites. Macromol. Mater. Eng. 2009;294:877–886.10.1002/mame.v294:12
- Feitosa VP, Ogliari FA, Van Meerbeek B, et al. Can the hydrophilicity of functional monomers affect chemical interaction? J. Dent. Res. 2014;93:201–206.10.1177/0022034513514587
- Catel Y, Schörpf S, Moszner N. Monomers for adhesive polymers, 16 synthesis of acidic dimethacrylates starting from thioglycerol. Macromol. Mater. Eng. 2015. doi:10.1002/mame.201500118.