
Abstract
The chemical composition of atmospheric particulate matter at Mt. Åreskutan, a mountaintop site in central Sweden, was analysed with a focus on its carbonaceous content. Filter samples taken during the Cloud and Aerosol Experiment at Åre (CAEsAR 2014) were analysed by means of a thermo-optical method and ion chromatography. Additionally, the particle light absorption and particle number size distribution measurements for the entire campaign were added to the analysis. Mean airborne concentrations of organic and elemental carbon during CAEsAR 2014 were OC = 0.85 0.80
g m
and EC = 0.06
0.06
g m
, respectively. Elemental to organic carbon ratios varied between EC/OC = 0.02 and 0.19. During the study a large wildfire occurred in Västmanland, Sweden, with the plume reaching our study site. This led to significant increases in OC and EC concentrations (OC = 3.04
0.03
g m
and EC = 0.24
0.00
g m
). The mean mass-specific absorption coefficient observed during the campaign was
= 9.1
7.3 m
g
(at wavelength
= 637 nm). In comparison to similarly remote European sites, Mt. Åreskutan experienced significantly lower carbonaceous aerosol loadings with a clear dominance of organic carbon. A mass closure study revealed a missing chemical mass fraction that likely originated from mineral dust. Potential regional source contributions of the carbonaceous aerosol were investigated using modelled air mass back trajectories. This source apportionment pointed to a correlation between high EC concentrations and air originating from continental Europe. Particles rich in organic carbon most often arrived from highly vegetated continental areas. However, marine regions were also a source of these aerosol particles. The source contributions derived during this study were compared to emission inventories of an Earth system model. This comparison highlighted a lack of OC and EC point-sources in the model’s emission inventory which could potentially lead to an underestimation of the carbonaceous aerosol reaching Mt. Åreskutan in the simulation of this Earth system model.
1. Introduction
Aerosol particles are an essential part of the Earth’s atmospheric system. They affect the planet’s radiative balance in two ways: directly as they scatter and absorb radiation and indirectly as they influence cloud properties. Presently, the uncertainty in direct and indirect aerosol effects on the climate dominates the overall uncertainties in radiative forcing estimates (IPCC, Citation2013). Besides its climatic effects, atmospheric particles are known to have adverse health effects on humans (UNEP/WMO, Citation2011).
On an annual basis, up to 40% of the total European aerosol mass is made up of carbon containing aerosol particles, referredto as total carbon (TC), with a significant portion consisting of organic matter [OM; ][]Yttri. The quantification of these carbon containing aerosol particles is based on bulk properties of carbonaceous compounds. Different bulk fractions are usually operationally classified as organic carbon (OC) and elemental carbon (EC).
Aerosol particles containing organic carbon are characterized by polyaromatic molecules and many other types of organic molecules that can contain polar functional groups. This variability in chemical composition can affect how particles take up water vapour, a property generally referred to as particle hygroscopicity (Brasseur et al., Citation2003). Direct sources of terrestrial OC include primary combustion processes like fossil fuel- or biomass-burning, while secondary organic aerosol particles originate from gas-to-particle conversion reactions of gaseous precursors. The precursor gases can be emitted from combustion processes or have natural sources such as emissions from vegetation.
A minor fraction of the TC mass consists of EC, more generally referred to as black carbon (BC). This fraction is non-hygroscopic, refractory and shows a graphite-like structure (Coz and Leck, Citation2011). Elemental carbon aerosol particles are mainly the result of incomplete combustion processes. Such processes can either be anthropogenic, as is the case with the burning of fossil fuels or the burning of biomass for forest clearing and cooking, or natural as is the case with forest fires. However, present-day EC originates mainly from anthropogenic sources.
Depending on the optical properties of the particulate matter and the albedo of the Earth’s surface, the interaction of aerosol particles with incoming solar radiation can lead to either a cooling or a warming of the atmosphere. EC absorbs light with high efficiency across the whole visible spectrum (Bond et al., Citation2013; Petzold et al., Citation2013) which mainly leads to a warming of the atmosphere (positive radiative forcing). Further, if EC is deposited on bright surfaces, like snow and ice, the change of surface albedo can potentially lead to faster melting rates and amplify the warming (Ramanathan and Carmichael, Citation2008). In contrast, for most OC types, the light scattering contribution dominates the light extinction which generally leads to an enhanced cooling effect of OC [negative radiative forcing;][]Fuzzi2006.
The generally short atmospheric residence times of aerosol particles containing a high fraction of organic carbon, alongside their highly variable chemical composition, morphology, mixing state and size, result in the potential for large regional differences. This makes the quantification of their regional climate and health effects very challenging. Therefore, additional research on ambient carbonaceous particulate matter is required at the local, regional and global scale (Brasseur et al., Citation2003). Monitoring emissions and their dispersion can help to improve climate models by providing new knowledge and new data for the validation of chemical transport models. To study the properties of aerosol particles and clouds in relation to different air mass regimes, the Cloud and Aerosol Experiment at Åre (CAEsAR 2014) was performed between June and October at a mountain site, Mt. Åreskutan, located in central Sweden. Measurements made during the campaign include the chemical characterization of airborne particulate matter (PM), precipitation samples and fog-water samples. For the purpose of a mass-closure study, carbonaceous and water-soluble inorganic aerosol components were quantified. Further, the mass-specific absorption coefficient was determined for the first time at Mt. Åreskutan. As such, this study complements previous monitoring observations for various stations across Europe as part of the European Monitoring and Evaluation Programme, EMEP (Tørseth et al., Citation2012). In this study, we focus on the carbonaceous components of ambient aerosol particles and we interpret our observations in the context of the various aerosol source regions influencing the chemical signature observed at Mt. Åreskutan. The measured carbonaceous aerosol fractions are placed into the context of data reported for similar sites across Europe and a comparison to an Earth system model is made in terms of the estimated aerosol source contributions.
2. Experimental
2.1. Measurement campaign and site
The Mt. Åreskutan station is located in a remote mountain region at 1250 m (a.s.l.) at almost the midpoint of Sweden (63 °26’N, 13 °6’E; see map in Fig. ). Air masses arriving at Mt. Åreskutan are considered to be representative of air arriving over central Scandinavia (Ogren and Rodhe, Citation1986). The Åre valley has approximately 1500 inhabitants and may be a source of aerosol particles at the mountaintop observatory station on occasion. A small sized restaurant located in the top station of the cable car presents the closest immediate anthropogenic emission source. The E14 road which connects the valley with Östersund (100 km east) and Trondheim, Norway (160 km west) is the only major road in the area.
Unlike many other comparable stations in Europe that present characteristic locations for certain air masses (polar, continental, marine or urban, etc.), a unique feature of Mt. Åreskutan is that it is located at the intersection of a number of air masses (Drewnick et al., Citation2007). This geographical setting makes it possible to study diverse air masses with varying degrees of anthropogenic influence at a single location.
2.2. In-situ measurements
2.2.1. Aerosol filter sampling
A low-volume reference sampler (LVS3 Small Filter Device, Leckel GmbH, Berlin, Germany) was used to collect PM with an aerodynamic diameter below 10 µm (PM) just outside the research station from 1:30 pm on 27 June until 1:30 pm on 13 October 2014. The instrument was operated at a flow rate of 2.3 m
h
for 72 h per sample. Aerosol samples were collected on micro-quartz fibre filters (12.07 cm
, T293, Munktell & Filtrak GmbH). Prior to sampling the filters were heated in a furnace at 800 °C for 12 h in order to remove organic compounds. Collected filter samples were each transferred to a clean petri slide and stored in the dark at +4 °C until further analysis. One part of the micro-quartz fibre filter was analysed for the OC and EC content using thermo-optical analysis, which is further explained in Sect. 2.3.1. Another part of each filter was extracted with water to determine the concentrations of water-soluble compounds further described in Sect. 2.3.2. Blank filters were loaded and unloaded into the reference sampler in the same manner as the sample filters and served as a reference.
2.2.2. Particle size distribution measurements
In order to determine particle size distributions in the sub-micron range, a differential mobility particle sizer (DMPS) consisting of a differential mobility analyser (DMA) and a condensation particle counter (CPC; TSI 3010) was used. The instrument was operated behind a PM inlet at a flow rate of 1 dm
min
. The ambient air was passed through a bipolar charger (neutralizer, Ni-63) before entering the closed-loop, custom-built DMPS. Here, the negatively charged particles were collected using positive high voltage in the DMA and subsequently counted with the CPC. Aerosol particles were measured within 40 size bins for particle mobility diameters
= 10–500 nm. Each size bin was measured for 10 s, with a lag time of 5 s introduced before measuring the next bin. The instruments’ sizing performance was verified with latex spheres of known size. Uncertainty in sizing for the latex spheres was found not to vary over size and determined to be
2 nm in diameter. The integrated number concentration calculated using the measured size distributions was compared to an independent CPC (TSI 3010) with a cut-off diameter of 10 nm. The instruments were operated in parallel and yielded agreement within 10% considering only aged aerosol particles (i.e. no new particle formation).
Particles with optical diameters up to 10 µm were measured with a white light aerosol optical particle size spectrometer (WELAS; PALAS GmbH, Model Promo 2000) operated at a sample flow of 5 dmmin
. The instrument measures light scattering on a single particle basis. A routine calibration with CalDust 1100, a calibration dust provided by the manufacturer with a refractive index of 1.43 and a diameter of 1.1 µm, was performed to account for any drifts in the instrument sensitivity. An empirical calibration using polystrene latex spheres (PSLs) with a certified size and a known scattering cross-section [for more details see e.g.][]Heim2008,Rosati2015 was used to relate the pulse height (voltage) to the scattering cross-section (particle property). For the optical analysis, particles were assumed to be spherical with an index of refraction of 1.59 (PSL).
It should be noted that all results presented in this study are given for conditions at ambient temperature and pressure.
2.2.3. Optical determination of equivalent black carbon
Real-time equivalent black carbon (eBC) mass concentrations were determined using a multi-angle absorption photometer (MAAP, Thermo Fisher Scientific Inc., Model 5012). The MAAP measures the intensity of light [ = 637 nm;][]Muller2011 transmitted through or backscattered from an aerosol loaded glass microfibre filter with subsequent determination of the aerosol absorption coefficient
. Full details of the algorithms used by the instrument, including the application of relevant correction factors, are given by Petzold and Schönlinner (Citation2004). Assuming BC is the dominant light-absorbing aerosol constituent, eBC mass concentrations were calculated by applying the mass-specific absorption coefficient
according to the following relationship:
(1)
For the initial determination of eBC concentrations was assumed to be
= 6.6 m
g
based on the findings of Petzold et al. (Citation2002). Note, that in an earlier study by Müller et al. (Citation2011), the MAAP was found to operate at a wavelength of 637 nm instead of 670 nm as stated by the manufacturer. Therefore, all values of
have been multiplied by a factor of 1.05 as recommended by Müller et al. (Citation2011).
2.3. Filter sample analysis
2.3.1. OC/EC thermo-optical analysis
In order to quantitatively divide the carbonaceous fraction of collected aerosol particles into OC and EC, a thermo-optical transmittance (TOT) analysis was applied. The method was first developed by Birch and Cary (Citation1996) and is described in detail by Wallén et al. (Citation2009). In brief, a section of 1.5 cm or, if particle loadings were high enough, 1 cm
of the three-day filter samples were transferred to a TOT carbon aerosol analysis instrument (Sunset Laboratory B.V., manufactured in 2003). The aerosol sample collected on the micro-quartz fibre filter was step-wise heated according to the temperature protocol EUSAAR-2 [European Supersites for Atmospheric Aerosol Research;][]Cavalli2010.
In the first analysis step, the measurement is carried out in a non-oxidizing environment where OC containing compounds evaporate and generate gaseous products. The latter are detected by means of flame ionization detection (FID) after oxidation to carbon dioxide followed by the catalytic conversion to methane. By introducing oxygen into the environment in the second step, the EC fraction of the sample combusts and gaseous combustion products are quantified in the same manner as before. Through the constant measurement of the filter transmittance any char formation can be corrected for. The point at which the transmittance recovers to its original value is referred to as the split point. Carbonaceous compounds measured before that point are defined as OC, while carbonaceous species evolved after the split point are assigned to the EC fraction. Towards the end of the analysis the entire carbon content, i.e. the TC loading, has volatilized and the FID is calibrated by introducing a known amount of methane. The previously unknown TC concentration of the sample can then be calculated.
A set of two to three filter sections was analysed for each sample. The maximum deviation of the different sections to the sample mean concentration was on average 4.6% for OC and 12.4% for EC, respectively. The lower detection limit of this method is on the order of 0.2 µg per cm filter for both OC and EC (Sunset Laboratory, Citation2010). For our sampling conditions this translates into concentrations of 0.01 µg per m
air. In order to evaluate the instrument performance and to standardize the methane and helium gas, 10 µL samples of 8 g L
sucrose were run regularly. This only served as a calibration agent for OC. A calibration for the refractory fraction was not performed since a suitable standard reference material consisting of pure EC does not exist (Baumgardner et al., Citation2012; Panteliadis et al., Citation2015).
Determining the split-point correctly is challenging. It depends not only on the thermal protocol and the associated difference in char formation, but also on the method used for the correction of char (transmission or reflectance) and the chemical properties of the aerosol (Karanasiou et al., Citation2015). This should be borne in mind when comparing reported OC and EC concentrations. It should also be noted that the concentrations of the single filter samples reported here are given as the sample average concentration and the uncertainty ranges represent the maximum deviation of the analysed filter sections to this average. This will further be referred to as sample mean variation in measurements.
2.3.2. Determination of the water-soluble mass fraction
Aqueous extracts of the ambient and blank filter samples were analysed for water-soluble ions by suppressed ion chromatography (IC). For extraction, a section of the micro-quartz fibre was put into a sterile plastic tube and 8 mL ultrapure water (MilliporeRios >18 M cm resistivity) was added. The tubes were closed and treated in an ultrasonic bath for 65 min to ensure all water-soluble ions were solved into solution. Subsequently the samples were stored in the dark at +4 °C until further analysis. Filtration of the extracts was not carried out.
Ion concentrations were determined using a Dionex ICS-2000 ion chromatography system (Thermo Scientific). Major anions were analysed with a Dionex AG11 pre-column and a Dionex AS11 main column. A Dionex CG16 pre-column and a Dionex CS16 main column were used for cation analysis. Before the injection valve (injection volume = 50 µL) the sample was freed from carbonates and other ionic contamination in a Dionex ATC-1 column. The measurement quality was assessed with certified internal and external reference samples [organized by][]ICreport2008. Systematic errors were 2% for all analysed ions (except for magnesium with
3%). Analytical detection limits, defined as twice the level of peak-to-peak instrument noise, were 0.2 µEq L
for ammonium, NH
, and between 0.00 and 0.05 µEq L
for all other constituents. Limits of quantification were defined as double the mean concentration determined on the blank filters.
The analysis of calcium ions, Ca, proved difficult for samples collected during this study since high concentrations were observed in the blank filter extracts. This, combined with the fact that low concentrations were observed in most samples, meant that Ca
was found to be below the limit of quantification for most samples. Therefore, Ca
concentrations were excluded from further analysis.
Of all samples, 25% showed excess negative ions of 50 µEq L or more. If the missing positive equivalents in these samples were assumed to the equal oxonium ions (H
O
), estimated pH-values ranged from 3.7 to 5.1 (mean pH = 4.4
0.6). Missing negative equivalents could be explained by the lack of carbonate measurements or organic ionic species that were not part of the analysis. Excess positive charge was only observed in a minor fraction of the samples and rarely exceeded 10 µEq L
.
3. Data processing and source analysis methods
The station on Mt. Åreskutan is generally within the planetary boundary layer (PBL) and frequently engulfed in clouds. However, for the trajectory source analysis we focus on the out-of-cloud periods to reduce the effect of recent local removal by precipitation.
There are different techniques available to measure carbonaceous particles. For example, an aerosol mass spectrometer (Q-AMS), has been used with this aim at Mt. Åreskutan previously (Drewnick et al., Citation2007). It provides a high time resolution of the aerosol’s OC fraction. A major disadvantage of this instrument is that it does not provide information about the refractory particles (EC), and therefore, a separate instrument is required. Our approach was to use the filter technique, based on the TOT analysis. Currently, all EC measurements are dependent on the analysis technique used, while TOT analysis with different instrumentation is known to give consistent TC results (Chiappini et al., Citation2014). Compared to more advanced systems this approach is considerably less cost- and labour-intensive. However, we needed to circumvent a major drawback, which is the coarse temporal resolution. Long sampling intervals were necessary since particularly clean air advected from polar or maritime areas was assumed to contain very low carbonaceous aerosol concentrations.
In order to obtain detectable concentrations of OC, EC, and inorganic ions on our filter substrates, we sampled for relatively long periods (72 h). With the aim of resolving potential variability in OC and EC during these periods two different proxy measurements were used for EC and OC, respectively. In the case of EC, hourly averages of the aerosol light absorption coefficient measured with the MAAP (reported every 10 s) served as a proxy. Hourly EC data, EC
(t), were obtained from the 72 h mean concentrations,
, by introducing the temporal variation of the proxy value
in the following manner:
(2)
where the variables b and
describe the 1h and the 72 h mean values of
measurements, respectively. This implies that
=
with t
, where EC
is the EC concentration at the i
hour (
) in a 72 h filter sample and t
and t
are the start and end times of the corresponding sampling period.
For the OC fraction, the sub-micrometer aerosol volume concentration derived from the measured aerosol particle size distributions was used. This is a reasonable assumption given that OC dominates in aerosol particles at Mt. Åreskutan (see Sect. 4.3) and because they occur mainly in the PM size-range (aerodynamic diameter
1 µm) at this site (Drewnick et al., Citation2007). Organic carbon concentrations for each hour of the campaign, OC
(t), were obtained from 72 h mean concentrations,
, with the help of 1h mean sub-micrometer volume concentrations V
and 72 h mean values
through a relationship equivalent to that shown in Eq. Equation2
(2) . Here, OC volume fractions were assumed to remain constant during the 72 h filter sampling intervals.
Table presents statistical parameters of the ratio of the respective proxies to the carbon fractions when averaged over a 72 h time window. The carbon fractions are clearly correlated to their respective proxies (see Fig. S2 in the supplementary material). Elemental carbon to OC ratios were calculated by dividing the respective concentrations. It should be noted that very low OC and EC concentrations were determined occasionally resulting in an implausibly large ratio. Therefore, only data points for which OC concentrations were larger than the 10th percentile of the data-set were taken into account for these calculations.
Following the transformation of three-day average OC and EC values into a more highly resolved hourly time series we proceeded to subdivide the data-set into out-of-cloud and in-cloud classes depending on the measured relative humidity (RH). Data were deemed to be out-of-cloud if hourly averaged RH 95% and liquid water content
0.05 g m
. Overall, 76% of the sampling hours corresponded to out-of-cloud data points.
3.1. Geographic source analysis
To study the potential sources of EC and OC at the station, we combined our out-of-cloud data points with modelled air parcel trajectories. Three-dimensional seven-day (168 h) back trajectories were calculated using the HYSPLIT (Hybrid Single-Particle Lagrangian Integrated Trajectory) model (Draxler and Hess, Citation1998; Draxler, Citation2004). The calculations are based on meteorological data from the NCEP’s (National Weather Service’s National Centres for Environmental Prediction) Global Data Assimilation System (GDAS). Each trajectory consists of 168 endpoints, one per hour. Air masses were modelled to arrive at altitudes of 1000 and 1500 m a.s.l. at the research stations coordinates for every hour of the campaign period. The seven-day back trajectories were used to study the potential contribution of EC and OC sources through the apportionment of the observed concentrations on a local grid using a separate cluster analysis. The choice of the trajectory length is based on estimates of the tropospheric lifetimes of BC and organic aerosol particles using the AeroCom models [][chap. 7]IPCC2013. Each trajectory was associated with the concentration value corresponding to the air masses arrival time at the station (see Eq. Equation2(2) ). This source analysis was not performed for the inorganic ions present in the water-soluble fraction of the aerosol. This was simply because no proxy for any of the inorganic ions with a high time resolution exists.
Table 1. Mean, standard deviation and percentile values of the ratio of the absorption coefficient to elemental carbon, EC, ( mass-specific absorption coefficient;
) and PM
volume to organic carbon, OC, for the three-day averages. See also Fig. S2 in the supplementary material.
3.1.1. Trajectory cluster analysis
Back trajectories of air masses arriving at 1000 m a.s.l. were grouped into trajectory clusters to see whether concentrations differed for air masses arriving from different direction sectors. Cluster analysis is a technique to separate a large data-set into groups with similar properties. The kmeans clustering algorithm (kmeans.m function available in MATLAB version 6.5 and later) was used. This function divides a data-set into a specified number of groups where observations within a group are closely related to each other and each group has the least possible similarities to the other groups. The technique uses the squared Euclidean distance measure and is described in more detail by Tunved et al. (Citation2004). As a result of this analysis, the trajectories were divided into seven clusters. Various numbers of clusters were tested but the differentiation into seven groups resulted in an optimal balance between the number of clusters and the difference in the characteristics of each cluster.
3.1.2. Potential source contribution
In addition to the trajectory cluster analysis, carbonaceous aerosol concentrations were apportioned to specific grid cells on a world map using the same approach as Zieger et al. (Citation2015). Back trajectories of both arrival altitudes were averaged on a grid with a horizontal resolution of 0.5°x 0.5°. Each grid cell that contained a seven-day back trajectory within the PBL was allocated the carbonaceous mass concentration observed at the receptor site for this trajectory (PBL data are included in the HYSPLIT model output). Following this analysis, the median concentration was calculated for all grid cells with at least five ‘hits’. It should be noted that the measured values of OC and EC were left unchanged for the whole length of the air mass trajectory. Therefore, removal processes like scavenging or deposition of aerosol particles along the trajectory were not taken into account.
3.2. Mass closure analysis
We have performed a mass closure using the measured concentrations of total carbon and the measured water-soluble inorganic mass. Since particle loadings were generally low, a gravimetric determination of the total aerosol mass was not possible. Instead, a three-day mean total aerosol mass for each filter sample was calculated using the particle size distribution measurements (cf. Sect. 2.2.2) by assuming an average particle density of = 1.5 g cm
. This estimate is in line with particle densities reported for summer aerosol particles at other remote sites like Hyytiälä, Finland (Kannosto et al., Citation2008), Jungfraujoch, Switzerland (Cozic et al., Citation2008) and Monte Cimone, Italy (Putaud et al., Citation2004). The number size distributions were converted to volume concentrations with the assumption of spherical particle shape. The DMPS and the WELAS particle size distributions were merged at
= 440 nm as the last two size bins measured by the DMPS were 446.7 and 501.2 nm, respectively, and the WELAS counting efficiency is optimal from approx. 400 nm upwards (see fig. 5 in Rosati et al., Citation2015). Mean number size distributions were, therefore, interpolated around this diameter.
Considering the fact that the two instruments use substantially different methods for particle size determination, the distributions compared well within the uncertainty at the size channels at which they were combined. Figure S1a in the supplementary material illustrates the number-, surface- and volume-size distributions observed throughout the study. During the study, there were a small number of periods with increased concentrations of large particles (super-micron) which had a significant effect on the estimated mean particle volume. For the purpose of the mass closure calculations we decided to only use the lowest 95 percentile of WELAS particle number counts. As such the number-size distributions measured by the WELAS (particles larger than 440 nm) were integrated and periods when this value was in the top 5% were discarded. This resulted in the distributions shown in Fig. S1b.
4. Results and discussion
4.1. Organic and elemental carbon
The time series of measured OC and EC mass concentrations and their ratio are displayed in Fig. . It can be seen that the carbonaceous aerosol concentrations were generally higher and more variable in the beginning of the campaign (Fig. a and b). Average concentrations for the entire campaign period were OC = 0.85 0.80
g m
and EC = 0.06
0.06 µg m
. The average ratio of elemental to organic carbon (EC/OC) was 0.08
0.04. In contrast to the concentrations of the single carbonaceous constituents, their mass ratio varied more towards the end of the campaign (see Fig. c). Several transient pollution plumes from central Europe as well as a pronounced fire plume emitted during a major wildfire in the area of Västmanland, Sweden, on 31 July arrived at the site during the campaign. These events were reflected in the chemical composition of the collected samples, i.e. high OC and EC concentrations were observed during 3 – 12 July, 2 – 5 August or 7 – 10 September (see Fig. a and b). Organic carbon concentrations were highest during the warm summer months. A maximum concentration of OC = 3.44
0.00 µg m
was observed during 9 and 12 July when air arrived from the boreal forest region and densely populated areas in northeastern Europe. The fire plume which arrived at Mt. Åreskutan after the wildfire outbreak in Västmanland on 31 July contributed to the second highest 72 h mean concentration (OC = 3.04
0.03 µg m
) between 2 - 5 August. The minimum value of OC = 0.06
0.01 µg m
was measured during 10 – 13 October when temperatures were low and maritime air masses reached the site, i.e. surface residence times over terrestrial ground was limited. Elemental carbon concentrations showed no distinct seasonal pattern and depended largely on the origin of the sampled air mass. The highest EC concentration was measured during the Swedish fire plume event between 2 – 5 August with EC = 0.24
0.00 µg m
. The filter sample for this period was influenced by both the wildfire plume and air masses with long residence times over densely populated regions in central Europe. Elemental carbon concentrations below the limit of detection were observed between 25 and 28 September. During this period air parcels had travelled almost exclusively over marine areas at high altitudes before reaching the site. The maximum EC to OC mass concentration ratio of 0.20
0.02 (sample mean
error calculated using error propagation) was observed during 20 –23 August. The filter samples collected during this period sampled air arriving from the north with PBL residence times over oil rigs located near the Norwegian coast.
Figure 1. Time series of (a) OC and (b) EC concentrations and (c) the EC to OC mass concentration ratio (EC/OC). Measured concentrations for all 72 h filter samples collected during the study are displayed as thick lines. A time series with hourly resolution, that was estimated with the help of proxy measurements, is displayed in panel (a) and (b) as thin lines. Note that no particle number size distribution data was available for the period between 1 – 9 July so no hourly variation could be introduced to the 72 h OC concentration data.
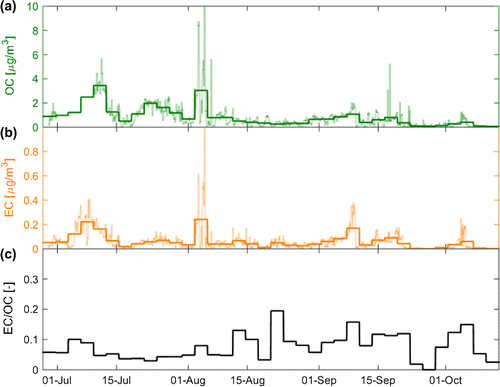
Figure 2. Carbonaceous aerosol concentrations for various sites in Europe. All sites resemble background locations similar to that at Mt. Åreskutan (see also Table ). The stations are ordered from South to North by latitude and are marked in the map in the upper right corner. The red star marks Mt. Åreskutan. Numbers above the bars are the carbonaceous mass concentration ratio (EC/OC). All concentration data were determined through thermo-optical transmittance analysis with either the NIOSH or the EUSAAR-2 temperature protocol (marked + or #, respectively). Red dates indicate that the study period overlapped with this study.
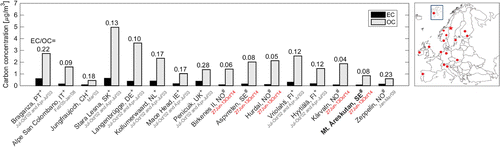
Figure compares our findings recorded during the campaign with measurements from similar remote sites in Europe. Reported concentrations and details on the sites are summarized in Table . All concentrations were obtained using the TOT method. In contrast to this study, most authors applied the NIOSH (National Institute for Occupational Safety and Health) protocol for the determination of EC and OC concentrations. Different protocols can result in substantially different concentrations. Several studies indicate a factor of two difference in EC concentrations when comparing the analysis of samples using EUSAAR-2 and NIOSH (Karanasiou et al., Citation2015; Bautista et al., Citation2015). The NIOSH protocol generally gives lower EC concentrations. As suggested by Panteliadis et al. (Citation2015), this is probably because the NIOSH protocol underestimates the amount of EC by interpreting it as OC. Almost no difference is observed in TC concentrations measured by the different protocols.
The comparison in Fig. suggests that the absolute mean concentrations of both EC and OC observed at Mt. Åreskutan during the campaign were relatively low. Elemental carbon concentrations reported for PM at the high alpine site Jungfrauchjoch in Switzerland in March 2004 are equally low. So were PM
EC concentrations reported for the Norwegian sites Birkenes II, Hurdal and Kårvatn for the period during which our campaign took place. Organic carbon containing aerosol particles constituted a rather large fraction of the total carbonaceous aerosol mass at Mt. Åreskutan compared to the other sites. Secondary organic matter generated through photochemical reactions of vapours emitted from the boreal forests on the Scandinavian peninsula is most likely responsible for this, as suggested by Drewnick et al. (Citation2007). The resulting low mass ratio of EC to OC is comparable to values reported for the other sites in Scandinavia (Aas et al., Citation2015; Yttri et al., Citation2007; Saarikoski et al., Citation2005). The low variability in the EC/OC mass ratio observed at Mt. Åreskutan is similar to the low variability observed at most of the Scandinavian sites (see Table ).
4.2. Determination of the mass absorption coefficient
An estimate for the mass-specific absorption coefficient at Mt. Åreskutan during the campaign was obtained by comparing the EC concentrations determined with the thermo-optical analysis with the aerosol light absorption coefficient
measured with the MAAP at
= 637 nm. This comparison assumes that EC is the dominant light absorbing aerosol component. The
for the absorption at 637 nm was calculated by dividing 72h mean values of the absorption coefficient
by the corresponding EC concentrations
. As shown in Table , a mean value of
= 9.1
7.3 m
g
was observed at Mt. Åreskutan during this campaign at
= 637 nm.
Reported -values for the same wavelength cover a large range (3.5 - 13.7 m
g
) as shown in a review by Bond and Bergstrom (Citation2006). The authors suggest an estimate of 7.5
1.2 m
g
for freshly emitted light absorbing carbon aerosol with decreasing values as the aerosol age increases as a result of coating by organic compounds. Processes such as this are likely to occur during the summer in remote regions such as Mt. Åreskutan due to enhanced photochemical activity. The value of
= 9.1 m
g
at
= 637 nm is in line with
-values reported for similar sites in Europe at the same wavelength. Cozic et al. (Citation2008) reported a value of 11.1
0.2 m
g
during the summer and 7.6
0.2 m
g
during the wintertime for the Jungfraujoch in Switzerland using the NIOSH temperature protocol (note, that they used major axis regression analysis, which would have resulted in a value of
= 10.5 m
g
if applied to our data; see Fig. S2a in the supplementary material). It should also be noted that the comparison to the values retrieved for the Jungfraujoch site have to be taken with caution since Cozic et al. used the NIOSH protocol. Zanatta et al. (Citation2016) recently reported summer values of 8.41 and 8.12 and fall values of 8.17 and 7.71 m
g
for the sites Aspvreten and Birkenes, respectively (geometric mean values) using the same EUSAAR protocol as in the present study. During our campaign, no clear difference could be seen in
for summer and fall.
4.3. Mass closure study
In order to test the consistency of the measurements, mass closure was attempted for the filter samples with the help of the measured carbonaceous aerosol fractions (measured by the TOT measurement) and inorganic ion constituents. Note, that water-soluble organic compounds measured by the ion chromatography are not included in the mass closure as this is already accounted for by the thermal-optical OC. To account for atoms other than carbon, OC concentrations were multiplied with an OM-to-OC-conversion-factor to obtain OM mass concentrations. Many different conversion factors have been reported in the literature and values between 1.6 and 2.1 are recommended (Turpin and Lim, Citation2001). Cozic et al. (Citation2008) reported a value of OM/OC = 1.84 for an alpine site with this value determined via the comparison of OC/EC-analysis and OM measurements from an aerosol mass-spectrometer. Malm et al. (Citation2005) reported a factor of approximately 1.8 when they attempted a mass closure in a US national park using fine mass measurements and light scattering coefficients. They observed that OM mass dominated the fine particle mass which is similar to our observations at Mt. Åreskutan (see below). Thus, we have used a OM/OC conversion factor of 1.8 during our study.
Table 2. Ambient EC and OC concentrations (meanstd) in PM
for various remote sites in Europe at different altitudes above sea level (a. s. l.). The sites are ordered from south to north by latitude and divided into the following categories: high alpine (Alp), Arctic (Arc) and rural background (RB). This comparison is illustrated in Fig. . Note that at Birkenes II, Apsvreten, Hurdal, Kårvatn, Mt. Åreskutan and Zeppelin the EUSAAR-2 temperature protocol was used, while at all other stations the NIOSH temperature protocol was used.
Figure a presents a time series of mass fractions for each of the components analysed on our filters. As a fraction of the total measured mass, the OM mass constituted between 20 and 84% with a campaign mean of 62%. In an earlier study at Mt. Åreskutan, Drewnick et al. (Citation2007) reported PM organic aerosol fractions between 49 and 86% depending on the source of the sampled air. In the same study, OM mass concentrations of above 80% were observed for air masses arriving at the site from Eastern Europe and via Finland. It is from these sources that we observed the highest carbon concentrations. Drewnick et al. (Citation2007) reported that SO
was responsible for 8 and 36% of the total mass analysed with the highest fractions observed for air masses reaching the site from the west, crossing the British Isles. During our campaign, SO
contributed a substantial fraction of between 9 and 69% (28% mean) to the total analysed PM
. The highest contributions were observed during the second half of the campaign (cf. Fig. a) when air masses arrived to the station predominantly with westerly winds. Mass fractions of NH
exhibited similar trends to SO
and dominated the water-soluble mass fraction (2 - 13%; 8% mean) together with SO
. The filter sample collected coincident to the passing of the Västmanland fire plume (2 - 5 August) showed the highest value of K
during the campaign (K
= 9.8
10
µg m
, contributing 1.3% to the analysed mass on this particular filter sample). Potassium has been used as a marker of forest fire smoke previously [e.g. ][]Sullivan2014. Indeed, the K
concentration determined from this specific filter is one order of magnitude larger than the campaign mean value of K
= 1.1
10
µg m
. Elemental carbon mass fractions were responsible for between 0 and 6% (2.5% mean) of the analysed mass.
Figure b presents the absolute mass of the measured components of each sample as well as the estimated mass for PM. The total aerosol mass reached a maximum between 2 and 5 August (9.11 µg m
) of which 7.76 µg m
could be explained by the mass of the analysed components. Aerosol loadings varied depending on meteorological conditions and air mass origin. On average, 78% of the total aerosol mass could be characterized with the applied methods (see Fig. ). The OM mass was responsible for a mean average of 48% (1.53
1.43 µg m
) of the total aerosol mass, while a mean average of 21% was composed of SO
(0.58
0.48 µg m
). The mean mass fraction of NH
was 6% (0.15
0.11 µg m
), while the mean mass fraction of EC was 2% (0.06
0.06 µg m
) during the campaign.
Figure 3. Time series of (a) the 72 h mean mass fractions of the total analysed mass and (b) the 72 h mean absolute concentrations of the measured aerosol components. The grey line in (b) shows the estimated total aerosol mass for PM derived from particle size measurements with the assumptions that all particles were spherical and that they had a mean aerosol density of 1.5 g cm
.
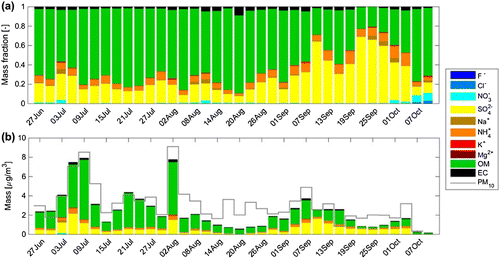
Figure 4. Mean mass fractions of the dominant aerosol components in PM measured using thermo-optical transmittance analysis and ion chormatography during CAEsAR 2014. The total aerosol mass was obtained by size distribution measurements assuming a mean aerosol density of 1.5 g cm
.
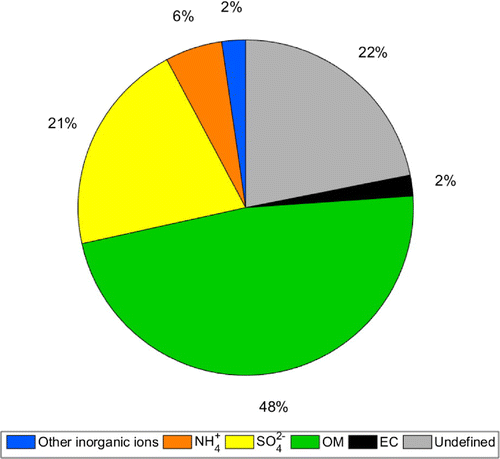
Figure 5. Average seven-day trajectories for the seven trajectory clusters are shown in (a). Black lines in the graphs around the main figure depict the standard deviation of all trajectories in each cluster to the average trajectory at each time step. The bar plot in (b) shows average carbonaceous aerosol concentrations with which the air masses of each trajectory cluster were associated during the study period (only cloud-free data points). The values in the figure depict the percentage of EC and OC relative to the TC concentration. Error bars show the standard deviation of the TC concentration data for each trajectory cluster (negative values are truncated). Values below the cluster names refer to the frequency with which trajectories in each cluster occurred during cloud-free periods.
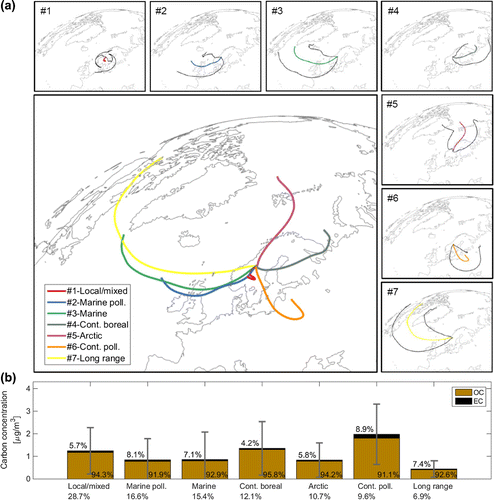
Table 3. Summary of the trajectory clusters and a description of the characteristics of air masses within each cluster. The analysis was based on hourly seven-day back trajectories calculated for the entire period of the study. Each of the trajectories terminated at 1000 m a.s.l. at the coordinates of the research station. The average elemental and organic carbon (EC and OC) concentrations with which trajectories of each cluster were associated are shown. The clusters are sorted according to the relative fraction of out-of-cloud trajectories (total fractional occurrence are shown in parenthesis) during the study period.
Overall, the average mass fractions for the compounds analysed during our study are consistent with the results derived with the Q-AMS by Drewnick et al. (Citation2007). This is despite the fact that although the average particle density of = 1.5 g cm
assumed in our study is in agreement with empirically derived mean
values for similar sites (see Sect. 3.2), it will not exactly match the particle density at Mt. Åreskutan and cannot account for temporal deviations. In addition to this, other assumptions highly influence estimates of total aerosol mass from particle size distribution measurements (Li et al., Citation2014; Szymanski et al., Citation2009). For example, ambient aerosol particles might have significantly different refractive indices and shape factors than the polystyrene latex spheres used for the calibration of the DMPS and WELAS.
Those periods characterized by large undefined mass fractions are likely to be influenced by particles composed of water-insoluble non-carbonaceous species such as mineral dust, which were not determined with the methods applied in our study. Mineral dust particles usually have relatively large diameters (> 1 µm); therefore, they can contribute substantially to particle volume and mass estimates. This is important to bear in mind given that Mt. Åreskutan is a popular tourist destination in summer, especially with mountain bikers. Activities such as this can contribute to an enhanced dust swirl close to the research station and therefore, lead to higher measured particle volume and mass. The mean size distribution measurements of DMPS and WELAS for the entire campaign revealed a significant coarse mode fraction of the particle volume-size distribution of 81% (see Fig. S1b).
4.4. Trajectory cluster analysis
The mean and standard deviation of the geographic origin of each of the seven clusters are shown in Fig. a. A description of the catchment areas of the different clusters represented is given in Table .
Air masses that circled close to the research station were grouped in the local/mixed trajectory cluster, #1. These air masses were observed most frequently during out-of-cloud periods and occurred during 29% of the study period. The transport of maritime air masses to the station could be subdivided into two types: one influenced by pollution sources in Northern central Europe travelling at low altitudes (#2 marine polluted; 17% occurrence), and one mostly influenced by marine aerosol particles arriving at the site from higher altitudes (#3 marine, 15% occurrence). Continental air masses occurred in two clusters: one in which trajectories covered boreal forest regions with considerable biogenic influence (#4 continental boreal; 12% occurrence), and one in which air masses travelled over densely populated, polluted areas (#6 continental polluted; 10% occurrence). Air parcels with trajectories in the Arctic cluster (#5) occurred during 11% of the study period. These trajectories originated over northern Scandinavia and northern marine regions. When passing over marine areas, air masses in this cluster might have been influenced by natural aerosol sources, but also by off-shore oil and gas production sites. About 7% of the analysed air was transported long distances across the Atlantic ocean (#7 long-range cluster).
The average concentrations of the carbonaceous aerosol particles for each of the trajectory clusters are shown in Table and illustrated in Fig. b. The highest mean TC concentration (1.981.33 µg m
) was measured for air originating in polluted continental regions followed by samples collected in air originating over the boreal region (1.36
1.18 µg m
) and the local/mixed cluster (1.25
1.03 µg m
). Aerosol particles with large EC to OC mass ratio were typical of the continental polluted and marine polluted trajectory clusters in which EC on average contributed 8.9 and 8.1% of the total carbonaceous aerosol particles, respectively. The lowest EC to OC mass ratios were observed in the continental boreal cluster where EC represented 4.2% of the total carbonaceous aerosol.
Figure 6. Results of the back trajectory analysis. Apportioned levels of the median (a) EC concentrations, (b) OC concentrations and (c) the EC/OC mass ratio measured at Mt. Åreskutan (red dot) during the study period. Grid cells were assigned concentration values of air masses analysed at Mt. Åreskutan if the air mass trajectory was located within the PBL at the location of the cell.
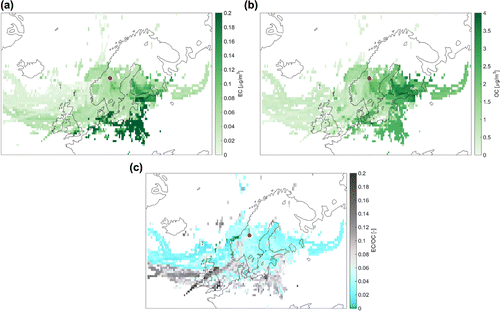
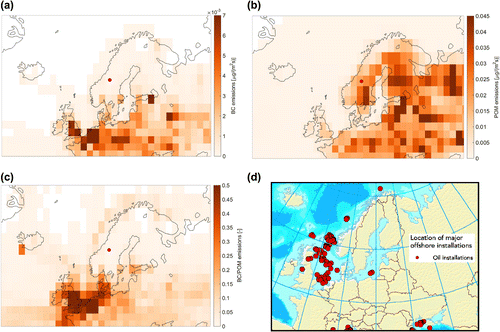
Figure 7. Aerosol emission fields used in the Norwegian Earth System Model (NorESM). Displayed are emission fields of (a) black carbon (BC), for which EC is the carbon-based proxy, (b) particulate organic matter (POM), which corresponds to OM and (c) the ratio of BC to POM emissions. The model emissions are mean present-day natural and anthropogenic emissions for the months of July, August and September. The figure in (d) shows major offshore oil installations (source: European Environment Agency; europa.eu).
4.5. Potential aerosol source contribution analysis
The regional source contribution was investigated by combining trajectory data with observations of carbonaceous aerosol particles. The method is intuitively simple and provides information on how much of a given quantity, on average, is observed at the receptor station (Mt. Åreskutan) after the air mass, as described by air-mass trajectories, has crossed a certain grid cell within the PBL. This provides a qualitative measure of the relative source contribution. However, as the method neglects parameters like particle loss processes, resulting maps provide an identification of apparent sources only. In addition, the general trajectory uncertainty increases with increasing trajectory time window (Gebhart et al., Citation2005). Nevertheless, it provides interesting insight into the distribution of regional emissions for those areas that frequently have connected flow with the receptor site.
The potential source analysis of the carbonaceous aerosol particles (median EC, OC concentration and the median mass ratio) collected during the study are shown in Fig. . High EC concentrations observed at the station are generally associated with sources in continental Europe. Source regions close to cities such as Prague, Tallinn or London are also visible (Fig. a). That relatively small-scale features like this can be identified using this method highlights the use of such an analysis.
The source signature for OC is more diverse, but one distinct feature is the strong contribution from the boreal forest region located to the east of the station (Fig. b). Interestingly, marine areas with off-shore oil and gas production sites also appear to be a notable source of OC (the location of major installations are shown in Fig. d).
As depicted in Fig. c, the EC/OC mass ratios exhibit a clear gradient from north to south; sources with higher EC/OC ratio are generally found over the continent of central Europe, while sources with lower EC/OC values and thus, higher organic contribution can be found over the oceans and the boreal areas. Further investigation is required to determine whether the sources with higher EC/OC mass ratios observed to the west of Ireland are ship traffic or other emission sources. For example, increased EC/OC mass ratios can also be caused by particles enriched in EC that were emitted at a later time and a location closer to our measurement site.
As mentioned above, no weighting of concentration values along a trajectory was used for the analysis. This may have led to apparent increases in concentration when trajectories associated with high carbon loadings were averaged in the same grid cell. It should also be noted that the white regions in Fig. do not necessarily indicate a lack of emission sources. Rather, they are the result of too little data in these grid cells to calculate a reasonable source strength (cf. Sect. 3.1). In this regard, continental central Europe might be expected to provide a significant source of carbonaceous aerosol. However, air masses originating in this region did not reach Mt. Åreskutan frequently during the campaign, as discussed in Sect. 4.4.
The potential source contribution maps shown in Fig. were compared with the emission fields used in a global climate model, in this case the Norwegian Earth System Model, NorESM (Bentsen et al., Citation2013; Iversen et al., Citation2013). The model includes estimations of surface emission quantities of various atmospheric components in the PBL at a horizontal resolution of 2° based on gridded emissions covering the historical period [1850–2000;][, Citation]. Emissions of BC, for which EC is the carbon-based proxy, and particulate organic matter (POM), which is comparable to OM, were calculated with NorESM for five years using the latest available emission data used in the Climate Model Intercomparison Project (Taylor et al. (Citation2012), 2006-2010). Maps of BC emissions, POM emissions and the BC/POM ratio are presented in Fig. . These emission fields and the maps generated following our source apportionment analysis (Fig. ) can only be compared qualitatively since the absolute magnitudes are not comparable. One obvious difference between the two is that the emissions in the model follow the contours of coastlines much more closely. This is understandable given the relatively limited amount of data collected during our study. There are also many similarities between the model emissions and our source apportionment analysis. For example, the high EC source regions of central Europe and southern Great Britain and the high OM originating in boreal forest regions are clearly visible in both.
Perhaps the most interesting aspect of this source apportionment is the relatively strong contribution from parts of the North Sea and Atlantic Ocean to the EC and OC observed at Mt. Åreskutan. We note that this particular region of the North Sea corresponds to the locations of major off shore oil exploration fields (see Fig. d). This clear source signal, to the organic carbon containing aerosol mass at the receptor site, was not corroborated by comparison to the emission fields used by the NorESM. The most likely reason is that fact that oil exploration in this region is not included in the emission inventory of the model.
Both our source apportionment and the model emission fields point to emissions of BC over the Atlantic. There is a large difference between the ratio of the carbonaceous fractions from our source apportionment fields when compared to the emission fields used in the model. In particular, differences are visible over the marine areas. One potential explanation for this is air pollution from ships that has been discussed for these regions by e.g. Aulinger et al. (Citation2016).
5. Summary and conclusions
Ambient aerosol particles collected at Mt. Åreskutan during the summer and fall of 2014 were analysed for their carbonaceous components and water-soluble constituents. A comparison to similar European monitoring sites suggests that although carbonaceous aerosol concentrations at Mt. Åreskutan were rather low they are comparable to other Scandinavian background sites. We observed low EC/OC mass ratios which underlines the influence of biogenic emissions on aerosol particles at Mt. Åreskutan.
We have attempted a mass closure which suggests that an average of 78% of the total aerosol mass could be characterized with the methods used during the study. Organic matter was identified as the major component of the aerosol particles sampled in terms of total mass, while the water-soluble aerosol mass was dominated by SO and NH
. We suspect that the mass fraction that remained undefined was composed of mineral dust.
A cluster analysis of the air mass trajectories arriving at the site suggested that the highest TC concentrations were transported from continental sites. Air transported from regions like Northern central Europe and the UK brought aerosol particles with enhanced EC to OC ratios to the site, while air advected over boreal forest regions transported aerosol particles with the lowest mean EC/OC values. The findings of our trajectory cluster analysis were supported by a source apportionment analysis.
Finally, comparison of the estimated sources of EC and OC observed at Mt. Åreskutan with emission fields used in the Norwegian Earth System Model suggests that off-shore oil and gas production sites may represent a significant source of OC containing aerosol particles to Mt. Åreskutan. This is significant because this aerosol source is currently not included in NorESM. We suggest that future updates to the emission fields in NorESM should include this missing source.
Supplementary Material
Download PDF (196 KB)Acknowledgements
We like to thank the Department of Environmental Science and Analytical Chemistry at Stockholm University for providing access to the Åreskutan sampling facility. We also thank Agneta Öhrström from the Department of Meteorology at Stockholm University for the conduction of the ion chromatographic analysis. We owe thanks to Lars ‘Lumpan’ Lundberg and the team at Skistar Åre for the extensive and enthusiastic support. The organic and elemental carbon data-sets for the Norwegian sites Birkenes, Hurdal and Kårvatn are obtained as part of the National monitoring programme funded by the Norwegian Environmental Agency. The data have been reported to the EMEP monitoring programme (Tørseth et al., Citation2012), and are available from the database infrastructure EBAS (http://ebas.nilu.no/) hosted at NILU. Last but certainly not least we also like to thank the helpers in the field, especially Birgitta Noone, Zahra Hamzavi, Tabea Henning, Matthias Tesche, Sebastian Arnoldt, Radek Krejci, Samuel Lowe, Therese Gadd, Peggy Achtert, Dan Partridge and Leif Bäcklin. The data collected during this study are available upon request from the authors.
Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
Supplemental data for this article can be accessed here: 10.1080/16000889.2017.1353387.
References
- Aas, W., Platt, S., Solberg, S. and Yttri, K. E. 2015. Monitoring of long-range transported air pollutants in Norway, annual report 2014. Tech. rep, NILU-Norwegian Institute for Air Research, P.O. Box 100, 2027 Kjeller.
- Aulinger, A., Matthias, V., Zeretzke, M., Bieser, J. Quante, M. and co-authors. 2016. The impact of shipping emissions on air pollution in the greater North Sea region-Part 1: Current emissions and concentrations. Atmos. Chem. Phys 16, 739–758.
- Baumgardner, D., Popovicheva, O., Allan, J., Bernardoni, V. Cao, J. and co-authors. 2012. Soot reference materials for instrument calibration and intercomparisons: a workshop summary with recommendations. Atmos. Meas. Tech. 5, 1869–1887.
- Bautista, A. T., Pabroa, P. C. B., Santos, F., Quirit, L. L. Asis, J. L. B. and co-authors. 2015. Intercomparison between NIOSH, IMPROVE-A, and EUSAAR-2 protocols: Finding an optimal thermal optical protocol for Philippines OC/EC samples. Atmos. Poll. Res. 6, 334–342.
- Bentsen, M., Bethke, I., Debernard, J. B., Iversen, T. and co-authors. 2013. The Norwegian earth system model, NorESM1-M-Part 1: Description and basic evaluation of the physical climate. Geosci. Model Dev., 6, 687–720.
- Birch, M. E. and Cary, R. A. 1996. Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust. Aerosol Sci. Technol. 25, 221–241.
- Bond, T. C. and Bergstrom, R. W. 2006. Light absorption by carbonaceous particles: An investigative review. Aerosol Sci. Technol. 40, 27–67.
- Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M. Berntsen, T. and co-authors. 2013. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 118, 5380–5552.
- Brasseur, G., Prinn, R. and Pszenny, A. 2003. Atmospheric Chemistry in a Changing World. Springer Verlag, Berlin, Heidelberg.
- Cavalli, F., Viana, M., Yttri, K. E., Genberg, J. and Putaud, J.-P. 2010. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: the EUSAAR protocol. Atmos. Meas. Tech. 3, 79–89.
- Chiappini, L., Verlhac, S., Aujay, R., Maenhaut, W., Putaud, J. P. and co-authors. 2014. Clues for a standardised thermal-optical protocol for the assessment of organic and elemental carbon within ambient air particulate matter. Atmos. Meas. Tech. 7, 1649–1661.
- Coz, E. and Leck, C. 2011. Morphology and state of mixture of atmospheric soot aggregates during the winter season over Southern Asia-a quantitative approach. Tellus B 63B, 107–116.
- Cozic, J., Verheggen, B., Weingartner, E., Crosier, J., Bower, K. N. and co-authors. 2008. Chemical composition of free tropospheric aerosol for PM1 and coarse mode at the high alpine site Jungfraujoch. Atmos. Chem. Phys. 8, 407–423.
- Draxler, R. R. 2004. Description of the HYSPLIT4 Modeling System. Air Resources Laboratory, Silver Spring, MD, NOAA Technical Memorandum ERL ARL-224 edn
- Draxler, R. R. and Hess, G. D. 1998. An overview of the HYSPLIT4 modelling system for trajectories, dispersion, and deposition. Aust. Met. Mag. 47, 295–308.
- Drewnick, F., Schneider, J., Hings, S. S., Hock, N., Noone, K. and co-authors. 2007. Measurement of ambient, interstitial, and residual aerosol particles on a mountaintop Site in Central Sweden using an aerosol mass spectrometer and a CVI. J. Atmos. Chem. 56, 1–12.
- EANET. 2008. Report of the inter-laboratory comparison project 2007. Tech. rep., Acid Deposition Monitoring Network in East Asia, Niigata, Japan. Online at: http://www.eanet.cc/product.html
- Fuzzi, S., Andreae, M. O., Huebert, B. J., Kulmala, M., Bond, T. C. and co-authors. 2006. Critical assessment of the current state of scientific knowledge, terminology, and research needs concerning the role of organic aerosols in the atmosphere, climate, and global change. Atmos. Chem. Phys. 6, 2017–2038.
- Gebhart, K. A., Schichtel, B. A. and Barna, M. G. 2005. Directional biases in back trajectories caused by model and input data. J. Air Waste Manage. Assoc. 55, 1649–1662.
- Heim, M., Mullins, B. J., Umhauer, H. and Kasper, G. 2008. Performance evaluation of three optical particle counters with an efficient ‘multimodal’ calibration method. J. Aerosol Sci. 12, 1019–1031.
- IPCC. 2013. Climate Change 2013: The Physical Basis: Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge, UK, 1535 pp.
- Iversen, T., Bentsen, M., Bethke, I.. Debernard, J. B. and co-authors. 2013. The Norwegian earth system model, NorESM1-M-Part 2: Climate response and scenario projections. Geosci. Model Dev. 6, 389–415.
- Kannosto, J., Virtanen, A., Lemmetty, M., Mäkelä, J. M., Keskinen, J. and co-authors. 2008. Mode resolved density of atmospheric aerosol particles. Atmos. Chem. Phys. 8, 5327–5337.
- Karanasiou, A., Minguillón, M. C., Viana, M., Alastuey, A., Putaud, J.-P. and co-authors. 2015. Thermal-optical analysis for the measurement of elemental carbon (EC) and organic carbon (OC) in ambient air a literature review. Atmos. Meas. Tech. Discuss. 8, 9649–971.
- Lamarque, J.-F., Bond, T. C., Eyring, V., Granier, C., Heil, A. and co-authors. 2010. Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: Methodology and application. Atmos. Chem. Phys. 10, 7017–7039.
- Leng, L., Zhang, T., Kleinman, L. and Zhu, W. 2007. Ordinary least square regression, orthogonal regression, geometric mean regression and their applications in aerosol science. J. Phys.: Conf. Ser. 78, 1–5.
- Li, M., You, R., Mulholland, G. and Zachariah, M. R. 2014. Development of a pulsed-field differential mobility analyzer: a method for measuring shape parameters for nonspherical particles. Aerosol Sci. Technol. 48, 20–30.
- Malm, W. C., Day, D. E., Carrico, C., Kreidenweis, S. M. Collett, J. L. and co-authors. 2005. Intercomparison and closure calculations using measurements of aerosol species and optical properties during the Yosemite aerosol characterization study. J. Geophys. Res.- Atmos. 110, 1–21.
- M{\"u}ller, T., Henzing, J. S., de Leeuw, G., Wiedensohler, A., Alastuey, A. and co-authors. 2011. Characterization and intercomparison of aerosol absorption photometers: Result of two intercomparison workshops. Atmos. Meas. Tech. 4, 245–268.
- Ogren, J. and Rodhe, H. 1986. Measurements of the chemical composition of cloudwater at a clean air site in central Scandinavia. Tellus B 38, 190–196.
- Panteliadis, P., Hafkenscheid, T., Cary, B., Diapouli, E., Fischer, A. and co-authors. 2015. ECOC comparison exercise with identical thermal protocols after temperature offset correction -- instrument diagnostics by in-depth evaluation of operational parameters. Atmos. Meas. Tech. 8, 779–792.
- Petzold, A. and Schönlinner, M. 2004. Multi-angle absorption photometry: a new method for the measurement of aerosol light absorption and atmospheric black carbon. J. Aerosol Sci. 35, 421–441.
- Petzold, A., Kramer, H. and Schönlinner, M. 2002. Continuous measurement of atmospheric black carbon using a multi-angle absorption photometer. Environ. Sci. & Pollut. Res. 4, 78–82.
- Petzold, A., Ogren, J. A., Fiebig, M., Laj, P., Li, S.-M. and co-authors. 2013. Recommendations for reporting black carbon measurements. Atmos. Chem. Phys. 13, 8365–8379.
- Putaud, J.-P., Dingenen, R. V., Dell’Acqua, A., Raes, F., Matta, E. and co-authors. 2004. Size-segregated aerosol mass closure and chemical composition in Monte Cimone (I) during MINATROC. Atmos. Chem. Phys. 4, 889–902.
- Ramanathan, V. and Carmichael, G. 2008. Global and regional climate changes due to black carbon. Nat. Geosc. 1, 221–227.
- Rosati, B., Wehrle, G., Gysel, M., Zieger, P., Baltensperger, U. and co-authors. 2015. The white-light humidified optical particle spectrometer (WHOPS) -- a novel airborne system to characterize aerosol hygroscopicity. Atmos. Meas. Tech. 8, 921–939.
- Saarikoski, S., M{\"a}kel{\"a}, T., Hillamo, R., Aalto, P. P., Kerminen, V.-M. and co-authors. 2005. Physico-chemical characterization and mass closure of size-segregated atmospheric aerosols in Hyyti{\"a}l{\"a}, Finland. Boreal Env. Res. 10, 385–400.
- Sandrini, S., Fuzzi, S., Piazzalunga, A., Prati, P., Bonasoni, P. and co-authors. 2014. Spatial and seasonal variability of carbonaceous aerosol across Italy. Atmos. Environ. 99, 587–598.
- Sullivan, A. P., May, A. A., Lee, T., McMeeking, G. R. Kreidenweis, S. M. and co-authors. 2014. Airborne characterization of smoke marker ratios from prescribed burning. Atmos. Chem. Phys. 14, 10535–10545.
- Sunset Laboratory. 2010. Sample analysis method for organic and elemental carbon aerosols. Tech. rep.. Online at: http://www.sunlab.com/wp-content/uploads/Sunset-Lab-Analysis-Method.pdf
- Szymanski, W. W., Nagy, A. and Czitrovszky, A. 2009. Optical particle spectrometry-problems and prospects. J. Quant. Spectrosc. Radiat. Transfer 110, 918–929.
- Taylor, K. E., Stouffer, R. J. and Meehl, G. A. 2012. An overview of CMIP5 and the experiment design. Am. Meteorol. Soc. 90, 485–498.
- Tørseth, K., Aas, W., Breivik, K., Raa, A. M. F., Fiebig, M. and co-authors. 2012. Introduction to the European Monitoring and Evaluation Programme (EMEP) and observed atmospheric composition change during 1972–2009. Atmos. Chem. Phys. 12, 5447–5481.
- Tunved, P., Ström, J. and Hansson, H.-C. 2004. An investigation of processes controlling the evolution of the boundary layer aerosol size distribution properties at the Swedish background station Aspvreten. Atmos. Chem. Phys. 4, 2581–2592.
- Turpin, B. and Lim, H. J. 2001. Species contributions to PM2.5 mass concentrations: Revisiting common assumptions for estimating organic mass. Aerosol Sci. Technol. 35, 602–610.
- UNEP/WMO. 2011. Integrated Assessment of Black Carbon and Tropospheric Ozone. Technical Report, UNEP and WMO, Geneva, Switzerland.
- Wallén, A., Lidén, G. and Hansson, H.-C. 2009. Measured elemental carbon by thermo-optical transmittance analysis in water-soluble extracts from diesel exhaust, woodsmoke, and ambient particulate samples. J. Occup. Environ. Hyg. 7, 35–45.
- Winiger, P., Andersson, A., Yttri, K. E., Tunved, P. and Gustafsson, O. 2015. Isotope-based source apportionment of EC aerosol particlesduring winter high-pollution events at the Zeppelin Observatory, Svalbard. Environ. Sci. Technol. 49, 11959–11966.
- Yttri, K. E., Aas, W., Bjerke, A., Cape, J. N., Cavalli, F. and co-authors. 2007. Elemental and organic carbon in PM10 a one year measurement campaign within the European Monitoring and Evaluation Programme EMEP. Atmos. Chem. Phys. 7, 5711–5725.
- Zanatta, M., Gysel, M., Bukowiecki, N., Mijller, T., Weingartner, E. and co-authors. 2016. A European aerosol phenomenology-5: Climatology of black carbon optical properties at 9 regional background sites across Europe. Atmos. Environ. 145, 346–364.
- Zieger, P., Aalto, P. P., Aaltonen, V., Aj, M., Backman, J. and co-authors. 2015. Low hygroscopic scattering enhancement of boreal aerosol and the implications for a columnar optical closure study. Atmos. Chem. Phys. 15, 7247–7267.