
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Aerosol particle optical properties were measured continuously between years 2013–2015 at the Marambio station in the Antarctic Peninsula. Annual cycles of particle scattering and absorption were studied and explained using measured particle chemical composition and the analysis of air mass transport patterns. The particle scattering was found elevated during the winter but the absorption did not show any clear annual cycle. The aerosol single scattering albedo at nm was on average 0.96
0.10, with a median of 0.99. Aerosol scattering Ångström exponent increased during summer, indicating an increasing fraction of fine mode particles. The aerosol was mainly composed of sea salt, sulphate and crustal soil minerals, and most of the particle mass were in the coarse mode. Both the particle absorption and scattering were increased during high wind speeds. This was explained by the dominance of the primary marine sea-spray and wind-blown soil dust sources. In contrast, the back-trajectory analysis suggested that long-range transport has only a minor role as a source of absorbing aerosol at the peninsula.
1. Introduction
Antarctica is the most isolated continent on the planet and therefore the most pristine. However, the region of the Antarctic Peninsula has experienced some drastic environmental changes in previous decades. Driven by changes in atmospheric and oceanic temperatures, the Antarctic ice sheets have been declining which in turn can disturb the global oceanic-atmospheric climate system (Vaughan, Citation2006; Wouters et al., Citation2015). While environmental changes in the peninsula have thus far been rather local, initiated by the polar stratospheric ozone loss and natural climate variability, the ongoing global climate change is suggested to trigger another heating period in the peninsula by the end of the century (Turner et al., Citation2016).
Atmospheric aerosol particles have a significant impact on the climate of the snow and ice covered Antarctica. Aerosol optical properties, such as scattering and absorption of light by the particles, determine the aerosol net effect on the surface and atmospheric temperature changes. Particle scattering and absorption depend on their atmospheric ‘ambient’ size and composition, and is thus closely tied with particle hygroscopic properties (e.g. Tang, Citation1996). The climate effect of aerosol particles depend on the fraction of particle light scattering to extinction (sum of light absorption and scattering) and the underlying surface. A highly reflective underlying surface such as snow or ice can amplify the aerosol heating impact. Thus, any changes in atmospheric light reflectance caused by aerosol particles can drastically modify the surface and atmospheric radiative balance (AMAP, Citation2011). Deposition of absorbing particles on snow and ice decreases the surface albedo, i.e. less reflection of solar radiation, and changes the snow properties further enhancing the snow melt (e.g. Hansen and Nazarenko, Citation2004). Particles may also modify climate via acting as cloud condensation nuclei (CCN). The particle ability to serve as a cloud or ice nuclei depends essentially on their size and chemical composition. CCN particles affect formation and lifetime of the clouds, as well as modify their optical properties. For a complete understanding on Antarctic climate variability, the aerosol seasonal and spatial characteristics and their main drivers are essential.
The Antarctic aerosol consists mainly of sulphate, sea salt and crustal mineral components, largely influenced by the Southern Ocean emissions (Shaw, Citation1988). The aerosol mass concentrations are the highest in coastal regions. Budhavant et al. (Citation2015) showed that an average (i.e. arithmetic mean throughout the text) (Particulate Mass
2.5
m aerodynamic equivalent diameter) and
(Particulate Mass
10
m aerodynamic equivalent diameter) concentrations during summer were 4.3 and 5.1
g m
, respectively, with main chemical species of sea salt, sulphate and some crustal components. Chaubey et al. (Citation2011) measured average total mass concentrations of 6.0 and 8.3
g m
at Larsemann Hills and Maitri stations in East Antarctica during a summer season. On the other side of the Antarctic coast, at McMurdo station, average
concentrations measured at two close-by locations were 3.4 and 4.1
g m
(Mazzera et al., Citation2001). In the Peninsula, fine (
2
m aerodynamic equivalent diameter) and coarse (2–10
m aerodynamic equivalent diameter) mode aerosol average mass concentrations were 2.4 and 4.4
g m
, respectively (Artaxo and Rabello, Citation1992).
The marine sea salt aerosol, by nature, is highly scattering and facilitates cloud formation as it contains high quantities of potentially good CCN (Murphy et al., Citation1998). These are particles with favourable size and hygroscopic properties, able to form cloud droplets at low supersaturations and have been observed to be abundant in Arctic and Antarctic marine environments (e.g. Asmi et al., Citation2010; Zieger et al., Citation2010). In Antarctica, the sea salt fraction of the total aerosol is dominant especially at the coastal sites with mass concentrations extending from some hundreds of ng m up to several
g m
and further increasing towards the Southern Ocean (Artaxo and Rabello, Citation1992; Wagenbach et al., Citation1998; Virkkula et al., Citation2006; Weller and Lampert, Citation2008a; Teinilä et al., Citation2014). Fresh sea salt is dominated by the coarse mode aerosol particles while aerosol ageing during long-range transport increases the concentrations in the sub-micron range (Teinilä et al., Citation2014). Towards the inland of the Antarctica, sea salt concentration decreases drastically (Teinilä et al., Citation2000; Virkkula et al., Citation2006). In Antarctic high plateaus and at the South Pole, sea salt concentration reaches its maximum in winter (Bodhaine et al., Citation1986; Jourdain et al., Citation2008) when passage of marine sea salt rich aerosol to inland is facilitated by the intensified atmospheric circulation and winter cyclonic storms (Bodhaine et al., Citation1986; Ito, Citation1989; Kaspari et al., Citation2005).
The aerosol chemical characteristics are reflected in the aerosol optical properties and further in their climatic impacts. Antarctic aerosols are typically highly scattering with a single scattering albedo (SSA) ranging from 0.95 to 1, the average value being around 0.99 as observed in Neumayer station (Weller and Lampert, Citation2008a; Weller et al., Citation2013). However, not many studies report the SSA for Antarctic aerosol and therefore it isnot possible to infer its wider spatial variability. At Neumayer, the SSA had a clear seasonal cycle with a minimum observed in summer. Aerosol light scattering coefficient was on average 3.2 and 4.8 Mm, in summer and winter, respectively (Weller and Lampert, Citation2008a). This is an order of magnitude higher than scattering measured at the South Pole in the early 80’s (average of 0.28 Mm
) (Bodhaine et al., Citation1986), consistent with the increasing sea salt concentration gradient towards the coast. However, the baseline (minimum) scattering coefficients showed remarkably similar temporal variability, and even values, at both sites (Fiebig et al., Citation2014). Aerosols at the coastal Neumayer station and at the South Pole also have a similar wavelength dependent scattering, decreasing with increasing wavelength, further suggesting a dominance of smaller, fine mode, particles. The intensified transport of sea salt aerosol towards the pole during winter is detected as a wintertime maximum in scattering values (Bodhaine et al., Citation1986).
A small, but significant fraction of Antarctic aerosol particles is absorbing. The lowest Antarctic atmospheric absorbing black carbon (BC) (or soot) particle mass concentrations have been measured at the South Pole (50 pg m – 5 ng m
) (Hansen et al., Citation1988). At coastal Antarctic sites an average BC concentration of 1 ng m
was measured at Halley (Wolff and Cachier, Citation1998), average of 1.6–2.1 ng m
at Neumayer (Weller et al., Citation2013), 8.3 ng m
at Ferraz in the Antarctic Peninsula (Pereira et al., Citation2006),
20 ng m
at McMurdo (Hansen et al., Citation2001), 13 and 75 ng m
in Larsemann Hills and Maitri (Chaubey et al., Citation2010), and a monthly average range of 2–9 ng m
at Syowa (Hara et al., Citation2008). Maximum BC concentrations are observed at all of these sites in summer (December–January) and in spring (October–November) and a minimum in autumn (April–May). The main source of absorbing aerosol has been suggested to be biomass burning and wild fires in South-America, Africa and Australia: a hypothesis supported by the observed seasonal variability in BC (Wolff and Cachier, Citation1998; Pereira et al., Citation2006; Hara et al., Citation2008; Fiebig et al., Citation2009; Weller et al., Citation2013). Analysis of source regions using chemical tracers and back-trajectories further points towards southern latitude continents as a primary source of Antarctic BC, and in particular the frequently occurring biomass burning events there (Wolff and Cachier, Citation1998; Pereira et al., Citation2006; Hara et al., Citation2008; Fiebig et al., Citation2009; Stohl and Sodemann, Citation2010; Bisiaux et al., Citation2012; Hu et al., Citation2013; Weller et al., Citation2013). However, other anthropogenic sources, such as the Southern Ocean marine vessels and various fossil fuel burning sources can impact the measured concentrations (Graf et al., Citation2010; Stohl and Sodemann, Citation2010). In the peninsula region, ship traffic was suggested to be the dominant source of BC during summer (Stohl and Sodemann, Citation2010). BC concentration has been seen to increase with wind speed, which was explained by either the intensified cyclonic activity or the descent of higher altitude air masses during strong katabatic winds (Hara et al., Citation2008; Weller et al., Citation2013). Several studies also address the pollutionoriginating from Antarctica itself as an additional source of BC (Hansen et al., Citation2001; Chaubey et al., Citation2010; Stohl and Sodemann, Citation2010).
Secondary particle sources are also important in the Antarctic (Asmi et al., Citation2010; Kyrö et al., Citation2013; Fiebig et al., Citation2014; Weller et al., Citation2015). Fiebig et al. (Citation2014) suggested that the baseline (minimum) of aerosol number, and of total scattering annual cycle is explained by photo-oxidation limited aerosol formation. The main source of secondary aerosol are the marine sulphur and organic precursor emissions (Asmi et al., Citation2010; Fiebig et al., Citation2014) but also marine iodine species (Atkinson et al., Citation2012) and inland biogenic sources have been suggested to play a role (Kyrö et al., Citation2013). Mass spectrometry measurements by Giordano et al. (Citation2017) showed that a significant fraction of fine mode aerosol number during the early spring transition period also originates from a yet unidentified source.
In light of the previous observations, the Southern Ocean primary and secondary emissions and some periodical long-range transport control the Antarctic aerosol properties. However, data are sparse and a full consensus is missing. Questions remain on the climatic implications of the aerosols residing over Antarctica and on their anticipated future trends, in particular in the view of the increasing anthropogenic and touristic activities in Antarctic. The human footprint has been increasing especially on the Antarctic Peninsula, where very few studies on aerosols have been conducted until today.
Our main goal is to present long-term aerosol data from the Antarctic Peninsula and to explain the observed particle optical properties and their inter-annual cycles using chemical and meteorological observations and transport modelling. Results will give more insight into the connections between the peninsula’s aerosol properties and their natural and anthropogenic drivers, and will assist in estimating Antarctic aerosol spatial and temporal trends, aerosol main sources and their climatic implications.
2. Methods
2.1. Site description
Measurements were conducted at the Argentinian Antarctic station Marambio (64 15
S; 56
38
W) at 198 m a.s.l. (Fig. ). Marambio is a year-round operating station located on an island at the tip of the Antarctic Peninsula. It serves primarily for various scientific projects but also acts as a logistic centre for transport flights to some surrounding stations in Antarctica. Most of the activities take place during the summer season but around 40–70 persons also remain during winter at the station. The soil type is permafrost with a light snow cover during most of the winter season. More information on the measurement site can be found in previous publications (e.g. Karhu et al., Citation2003; Meinander et al., Citation2016).
Figure 1. Location of base Marambio on an island at the tip of the Antarctic Peninsula. On the right hand side is presented the wind rose calculated for years 2013–2015, showing frequencies of prevailing wind directions and wind speed in colours.
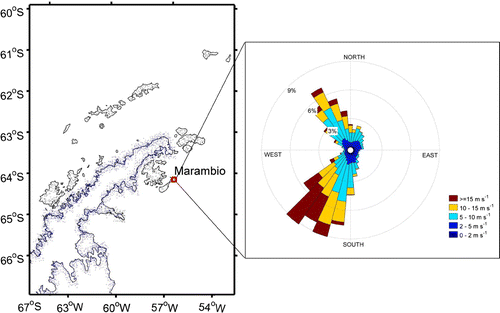
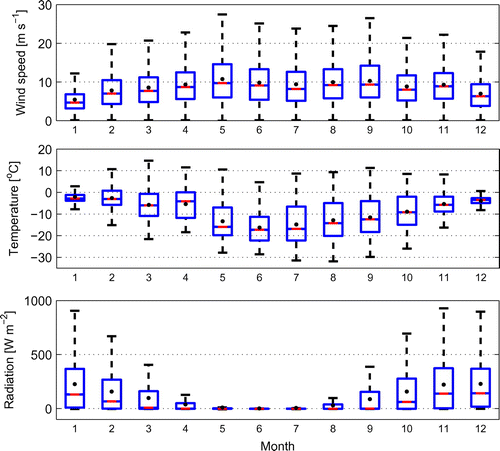
Figure 2. Wind speed, temperature and global radiation measured in Marambio during 2013–2015. Blue bars present the quartiles and whiskers the extremes in data for each month. Horizontal red lines depict the median and black dots the average values.
A specific laboratory container for aerosol measurements was transported and installed in Marambio in January 2013. The container is located about 300 m distance to the southwest from the main base and power generators. This location was selected to avoid local contamination, when the prevailing wind directions at the site are southwest and northwest (Fig. ). Occasional pollution, especially during the summer, may arise from activities at the airfield, located at about 500 m northwest from the container.
2.2. Measurement set-up
Aerosol measurements in Marambio started on 7 February 2013 and they have been running continuously since then. Here data until 30 November 2015 are presented and analyzed.
The sample air is drawn into the measurement container via a stainless steel inlet tube of 16 mm diameter. The inlet extends to a height of 6 m above the ground and 2 m above the roof of the container and is equipped with a Digitel Enviro-sense PM particle pre-separator with a nominal flow rate of 2.3 m
h
. Inside the container this flow is divided to different instruments via a custom-made flow-divider unit designed for isokinetic sampling with minimal particle losses. Sample lines are built direct from the roof to avoid larger particle aerodynamic losses and thus, no loss corrections are applied. Also no aerosol dryer is needed because of the indoor–outdoor temperature difference. The sample RH remained
30% throughout the year. Operation of instrumentation requires only one weekly maintenance visit during normal conditions.
2.3. Aerosol optical measurements
Aerosol particle scattering is measured with an Ecotech Aurora model 3000 nephelometer. It measures total aerosol light scattering between angles of 10–171
at the blue, green and red wavelengths (
) of 450, 525 and 635 nm (Müller et al., Citation2011) with a 60 s detection limit of
0.3 Mm
based on manufacturer. The nephelometer is run with an external pump where a flow rate of 5 L min
is maintained by a critical orifice. The correct flow rate is checked weekly. Scattering zero values are corrected based on automated daily filtered-air measurements during a 10-min period of which the last 5-min values are used for data correction. Every 2–3 months the nephelometer is calibrated with
gas. Calibration remained relatively stable during years 2013–2015, and if a significant deviation from previous calibration values was observed, calibration was repeated. Calibration line M (slope) values as calculated based on all the successful calibrations were:
,
, and
, for red, green and blue wavelengths, and calibration C (zero) values were:
m,
m, and 5.3
0.1 x 10
m, respectively. Data are corrected for truncation and angular non-idealities as suggested by Müller et al. (Citation2011) using the correction factors for no pre-cut option. Instrument time resolution is set to 1-min and averaged to 5-min or longer during data analysis. The high-resolution (1-min) data are used solely for cleaning of local contamination episodes and for data correction.
Aerosol particle absorption is measured with a Thermo Scientific Multi-angle absorption photometer (MAAP) model 5012. MAAP measures absorption at a wavelength of 637 nm (Müller et al., Citation2011) and reports an equivalent black carbon (EBC) mass concentration using a mass absorption cross-section (MAC) of 6.6 m g
. In the data analysis, both the EBC and the calculated absorption coefficients are used. EBC was taken direct from instrument given value, without the suggested 5% correction due to wavelength shift (Müller et al., Citation2011). MAAP flow rate is set to 15 L min
and validated every 2–3 months. Because of the low EBC concentrations at the site, no accumulation artefact correction was applied to the data (Hyvärinen et al., Citation2013). Time resolution is set to 1-min but similar to nephelometer data, these high-resolution (i.e. 1-min) values are used only for cleaning while for the analysis the data are averaged to 5-min or more. MAAP lower detection limit at 30-min resolution is 20 ng m
, which is high for Antarctica. For this reason data are often noisy.
Both aerosol scattering and absorption data are cleaned from local contamination. Data measured during winds from the station sector (0–90) or during wind speeds
1 m s
, are always removed. Additionally, data during weekly maintenance visits are removed automatically, because flow rate measurements and filter changes cause an opening of the inlet line and possible entrance of container interior air. Finally this pre-cleaned data is manually checked and any occasional short-term peaks or clearly erroneous values are removed from the data. Further averaging is done using this cleaned data-set.
In addition to local contamination, episodes of fog and low-level clouds could impact our aerosol results. Activated 10
m cloud droplet particles are not adequately collected in
inlet leading to an underestimation of the particle concentration. While this is recognized as a possible negative artifact, we did not monitor the episodes of fog or clouds and were therefore unable to remove any data affected by these. These episodes in Marambio are known to be more frequent during summer but firm statistics of them are missing. While they might distort the measured inter-annual cycles of aerosol parameters, we used relative humidity (RH) as a proxy for cloud and fog episodes and tested their impact. With various RH limits applied we did not, however, detect any significant systematic change in aerosol scattering parameter mean values nor in their inter-annual cycles.
Data coverage for both scattering and absorption between 7 February 2013 and 30 November 2015 was 100% and after cleaning the remaining 81% of the data were used for the analysis.
2.4. Chemical ion measurements and analysis
Aerosol samples for chemical analysis are collected using a virtual impactor (VI, Loo and Cork, Citation1988). The VI collects particles in two size fractions: fine (Dm) and coarse (2.5
m
D
m). Flow rate of the VI is 16.7 L min
, of which 15 L min
is used to collect the fine particles and the remaining 1.7 L min
is used for collection of the coarse particles. Particles are collected on 47-mm Teflon filters (Gelman Sciences, pore size 1
m). Fine filters are also weighted before and after collection with a Mettler-Toledo UMT2 Ultra Microbalance which has a precision of 0.1
g. Relative humidity of the filters during weighting is kept at 45–55%. This variability in RH can have a significant impact on hygroscopic aerosol mass (up to a factor of 1.4–2.5 for sea salt Tang et al., Citation1997), and is thus recognized as an important source of uncertainty regarding total measured particle mass. Collection time per filter is typically one week, but varied here between 5 and 12 days. Collected filters are stored on petri slides and kept in a freezer (
C) until analyzed. A blank is taken every 2–3 months by keeping the blank filter in open air during the handling of collected VI filters, and is treated otherwise in similar manner as collection filters. Collection is automatically stopped when winds are from the station sector (0–90
) as well as during wind speeds
1 m s
. A total of 124 coarse and 124 fine filters were collected between 7 February 2013 and 22 October 2015, which were analyzed and results presented here. Blank filters were collected five times per year, and a total of 15 for the whole time period considered here.
Filters were analyzed in an FMI clean room facility. The mass concentrations of sodium (Na), ammonium (
), potassium (
), magnesium (
), calcium (
), chloride (
), nitrate (
), sulphate (
), MSA (
) and oxalate (
) were determined from the filter substrates. Cation and anion analyses were done simultaneously with two ion chromatography systems (Dionex ICS-2000). Anions and cations were analyzed using AG11/AS11 and CG12A/CS12A columns, respectively. Detection of ions was done using conductivity detector. The lowest detection limit of this system for most ions is 1 ng mL
but is slightly increased, 3 ng mL
, for
and
ions. Filters were extracted in 5 mL of Milli-Q water with 10-min gentle rotation just before chemical analyzes.
The filter ion masses were converted into atmospheric concentrations using the measured flow rate and collection time for each filter. Average blank filter ion mass was subtracted from each collected filter ion mass. Blank correction was also made for weighted filter mass. Coarse fraction concentrations were corrected using the fine fraction concentrations. This correction is due to the fact that in VI 10% of the fine particles are collected on the coarse filter. Total aerosol mass was determined gravimetrically. Blank concentrations were subtracted from measured concentrations. The average blank concentrations were 5% of the average collected filter concentrations for most ions,
1% for chlorine, sulphate, sodium and magnesium, which were the most abundant ions and 8% for oxalate and 16% for nitrate, which in contrast represented very minor fractions of the total ion mass.
Chemical ion mass balance was calculated as in Virkkula et al. (Citation2006) and Asmi et al. (Citation2010). Sulphuric acid was only neutralized by ammonia and divided between sulphuric acid, ammonium bisulphate and ammonium sulphate based on equivalent molar ratio of . When this ratio was
1, aerosol was considered to be a mixture of H
SO
and (
, and when it was
2 all the sulphate was in the form of (
with some excess ammonia. Sea salt mass concentration was calculated based on both sodium and chlorine concentration as
. Soil mass was calculated as
. Non-sea-salt potassium, magnesium and calcium concentrations were calculated based on sodium concentration similar to Virkkula et al. (Citation2006).
2.5. Auxiliary measurements
A weather station has been installed on the roof of the measurement container. It consists of a Vaisala QML201L data logger connected with a Vaisala HMP155D humidity and temperature probe, Vaisala PTB220 pressure transmitter and a Thies 2D ultrasonic anemometer for wind speed and direction. Winds, temperature and relative humidity are measured at 10 m above the surface. Wind speed and direction data from this station are also used for automated filter collection control, mentioned above. Global radiation is measured with a Kipp&Zonen CMP11 pyranometer which is equipped with a blower and also manually cleaned weekly. All weather parameters are recorded as 1-min averaged values.
In addition, samples for Pb analysis were collected continuously between 18 March and 19 September 2013, altogether 59 samples.
Pb is formed in the atmosphere from 222-Rn and as such serves as a tracer for continental influence in air masses. High-volume (120 m
h
) aerosol particle samples were collected onto glass-fiber filters (Munktell MGA) and a new filter was exchanged every 2–5 days. The concentration of the
Pb on these aerosol filters was analyzed using the alpha counting of the in-grown daughter nuclide polonium-210 with an automatic alpha/beta analyzer (Mattsson et al., Citation1996).
2.6. Air mass back-trajectories
Air mass back trajectories were calculated using the HYSPLIT 4.9 model (Draxler and Hess, Citation1997; draxler98; draxler99. The back trajectory calculations were computed using the ensemble method, and the arrival height was 250 m above ground level. The ensemble method will produce 27 ensemble members for each back trajectory run by varying the meteorology in the horizontal and the vertical to account for uncertainties in the model input data. Input meteorological data for the HYSPLIT model runs were taken from the National Weather Service’s National Centers for Environmental Prediction (NCEP) Global Data Assimilation System (GDAS). Trajectories were calculated in 3-hour intervals starting at 00 UTC time with a total backward calculation time of 168-hours for each trajectory. The meteorological data provided were used to define the boundary layer height (BL) and only those trajectories traversing within this BL were considered to contribute as an aerosol source and were accounted for in the residence time calculation.
3. Results
3.1. Meteorological conditions
The weather in Marambio follows a seasonal pattern with lower temperatures and stronger winds in winter than in summer (Fig. ). Average temperature extended from about C in the winter to
C in January, with a range of values measured between
–10
C. Wind speed reached values of around 25 m s
during winter storms while the monthly averages were between 5–10 m s
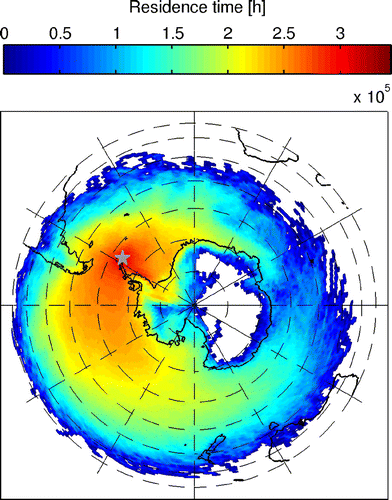
. Global radiation monthly averages mainly follow the changes between polar night and midnight sun, while in fact the cloudiness in Marambio is more frequent in summer than in winter (based on weather observations at the station). Prevailing wind directions are northwest (NW) and southwest (SW), but most storms occur at south-westerly winds (Fig. ). The air circulation follows the westerlies around the Antarctica where the Southern Ocean is the main source region (Fig. ). Indeed, marine open water regions contribute to 59.3% in total, with the second biggest contributor being the sea ice (34.8%). During local SW winds air mass origin remains marine but is circulating back towards the ocean following the eastern coast of the Peninsula. This circulation is likely to transport emissions from the Peninsula to the measurement station. Based on calculated back-trajectories, South-America is not a significant source region (other continents representing 0.8% of total contribution), but at times the westerlies move slightly towards the north. Then the southern tip of South-America can be considered a likely source region given its close proximity and wind patterns, and is likely to hold a strong anthropogenic signal. It should also be noted that Fig. comprises back-trajectories spanning seven days back in time and thus the source areas which are further away are not captured. In contrast, air from the central parts of Antarctica is practically never transported to Marambio.
Figure 3. Combined air mass back-trajectories for the measurements between 7 February 2013 and 30 November 2015. The trajectories were calculated using the ensemble method of HYSPLIT 4.9 and an arrival height of 250 m. The colour scale presents the total surface residence time and only times when trajectories were traversing within the boundary layer during the preceding 168 h were considered in the analysis. The residence time over different surfaces inside the PBL prior to arriving at the station were Antarctica (2.8%), other continents (0.8%), sea ice (34.8%), ice shelves (2.3%) and open water (59.3%).
3.2. Aerosol optical properties
The aerosol optical properties discussed in this section are summarized in Table . The monthly average aerosol scattering coefficients () at wavelengths
nm, 525 nm and 635 nm, respectively, extended from their minimum values of 2.5, 2.2 and 1.8 Mm
in December to their maxima of 7.2, 7.2 and 7.0 in June (Fig. a). The seasonal cycle of
had a minimum in summer and maximum in winter with a secondary maximum in spring (September–October). The average scattering coefficients are clearly influenced by some extreme events, while the monthly medians are always 20–50% below the average values. The overall average
was 5.1, 5.0 and 4.8 Mm
at
nm, 525 nm and 635 nm, respectively, and the average absorption coefficient
at
nm was 0.11 Mm
(Table ). The SSA at
nm was on average 0.96, with a median value of 0.99. This means that the majority of the aerosol particles are scattering rather than absorbing. The very weak wavelength dependence in the
further suggests that a large fraction of the scattering is due to coarse mode aerosol. Higher aerosol scattering during winter is in agreement with increasing marine aerosol production during stronger winds.
Table 1. Average standard deviation and median values of aerosol scattering coefficients, absorption coefficient, SSA, scattering SAE and equivalent black carbon concentration calculated for different wavelengths (presented in 2nd column) using hourly averages of data measured during period February 2013–30 November 2015.
Figure 4. (a) Scattering coefficient for red, green and blue wavelengths in representative colours, (b) EBC concentration, (c) SSA for 637 nm wavelength and (d) Scattering SAE for different wavelength regions. Bars present the quartiles and whiskers the extremes in data for each month during years 2013–2015. Horizontal lines depict the median and dots the average values.
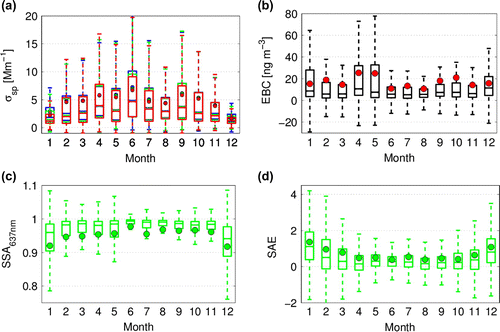
The overall average EBC concentration was 17.3 ng m and the median EBC was 6.9 ng m
(Table ). The lowest average EBC values were observed in winter from June to August: 11.0, 13.4 and 10.8 ng m
and the highest values of 25.4 and 25.0 ng m
were observed in April and May (Fig. b). However, the median EBC concentration does not show a strong seasonal cycle with values ranging between 5.3 and 11.5 ng m
, for all months. Similar to aerosol scattering, the large differences observed between median and average EBC values imply that the averages were influenced by extreme events, while the median values are most representative to the typical aerosol characteristics.
Table 2. Average concentrations [ng m] of fine mode fractions of nss-potassium (nss-
), nss-magnesium (nss-
), nss-calcium (nss-
), nss-sulphate (nss-
), methanesulfonic acid (MSA), nitrate (NO
), oxalate, ammonia (NH
) and sea salt, respectively, during October–March (summer) and April–September (winter) seasons for filter collections with average wind speed
10 or 12 m s
and
10 or 12 m s
, as well as concentrations calculated using all available data, with number of samples (n) used presented in the last row of the table.
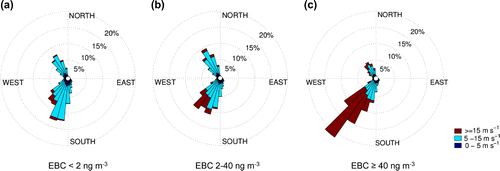
Figure 5. Wind roses for different EBC concentration percentiles with wind speed indicated in colours. Equivalent BC (15-min average) was used to separate the data into 3 groups: (a) low EBC (below 10th percentile), (b) medium EBC (between 10th and 90th percentiles) and (c) high EBC (above 90th percentile). Percentile limits of 10 and 90 correspond to EBC concentrations of 2 ng m and 40 ng m
, respectively.
The annual behaviour of the SSA is shown in Fig. c. The lowest monthly average values were observed in summer (December–January: 0.92). The highest average (0.98) and median (0.99) SSA values were observed in June when the aerosol scattering reached its maximum. The summer minimum in SSA indicates increasing anthropogenic activities at the station and in the region of the Antarctic Peninsula. The origin of the high EBC during April–May remains unexplained. The winter maximum in SSA could be a consequence of minimal local impacts and long-range transport. The SSA values above unity lack physical meaning and reflect that the instruments sometimes operate close to their detection limit.
Table 3. Average concentrations [ng m] of coarse mode fractions of nss-potassium (nss-
), nss-magnesium (nss-
), nss-calcium (nss-
), nss-sulphate (nss-
), methanesulphonic acid (MSA), nitrate (NO
), oxalate, ammonia (NH
) and sea salt, respectively, during October–March (summer) and April–September (winter) seasons for filter collections with average wind speed
10 or 12 m s
and
10 or 12 m s
, as well as concentrations calculated using all available data, with number of samples (n) used presented in the last row of the table.
Aerosol optical mean size was qualitatively examined using a scattering Ångström exponent (SAE), defined as(1)
(1)
where is the aerosol scattering coefficient at a wavelength
. Here, we used for smaller
= 450 nm and for larger
nm to calculate the aerosol SAE and obtained an annual average value of 0.6 (Table ). While the SAE describes the wavelength dependence of aerosol scattering, values below one typically indicate dominance of coarse mode aerosols, i.e. a large particle optical mean size. The highest median values of the SAE of around one were observed during peak summer months (December–January) while during the remaining of the year the median SAE typically stayed between 0.2–0.4 (Fig. d). These relatively low SAE values provide evidence of the dominance of the coarse mode aerosol during most of the year. The higher observed SAE in summer, in contrast, is indicative of additional fresh emissions, and possibly secondary particle formation, during this season. In winter when the particle scattering coefficients are the highest, also SAE decreases, indicating that scattering is dominated by coarse mode aerosol. This could be marine sea salt or soil re-suspension during strong winter storms. The origin of the aerosol will be examined further in following sections.
3.3. Aerosol source regions
The wind roses plotted for the lowest (10th percentile) and the highest (
90th percentile) EBC levels separately reveal some systematic changes in winds (Fig. ). The EBC concentration increases with wind speed, which is favoured during SW air flow. This would imply a connection with local emissions from the peninsula rather than with long-range transport as the main origin of EBC. Back-trajectories, showing residence time in the boundary layer, were equally plotted for different EBC levels separately, but showing no significant differences between them (Fig. S1). Relative contribution of different land types: Antarctica, other continents, sea ice, ice shelves and open water, also remained quite similar with changing EBC concentration (Table S1). Most air masses are marine, with high (around 60%) open water area contribution, and the second important source area type is sea ice. Those air masses with the highest EBC had the most influence from the sea ice and the least from open water. This might be related with SW air flow pattern from the eastern side of the peninsula, which is a region of a year-round ice cover. Interestingly, South-America does not represent a more significant source region for high than for low EBC data while the source contribution from other continents remains between 0.7–1.0% at all EBC ranges. This suggests that biomass burning and other anthropogenic sources in South-America are not major sources of absorbing aerosol in Marambio. Also the
Pb analysis made for the year 2013 showed a very minor continental influence (Fig. S2). Concentrations of active
Pb were typically below the detection limit (2
Bq m
), with a maximum of 6.9
Bq m
observed in the end of August 2013. In contrast to EBC wind roses, continental influences based on
Pb were mainly encountered during NW winds (Fig. S2), giving additional evidence of the minor role of South-American emissions here.
Figure 6. EBC concentration as a function of scattering coefficient for red (635 nm) wavelength. Circles present averaged data during different winter April–September (blue circles) and summer October–March (red circles) periods divided into five categories based on average wind speed. Circle size increases with wind speed as: 5 m s
, 5–10 m s
, 10–15 m s
, 15–20 m s
,
20 m s
. The grey colour scale within the circles presents the average scattering SAE. Standard deviation of EBC and scattering values are presented as red and blue lines. Quadratic fits for winter and summer data are presented with dashed lines of corresponding colour. Lines presenting SSA of 0.95, 0.97 and 0.99 are also plotted using grey and black colours to guide the eye and the average EBC concentration line for reference.
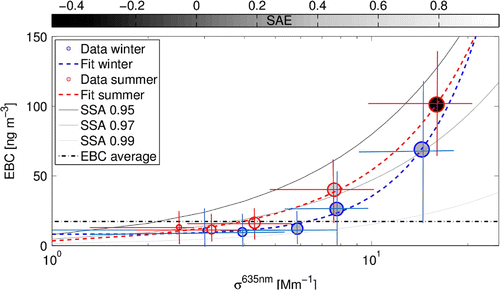
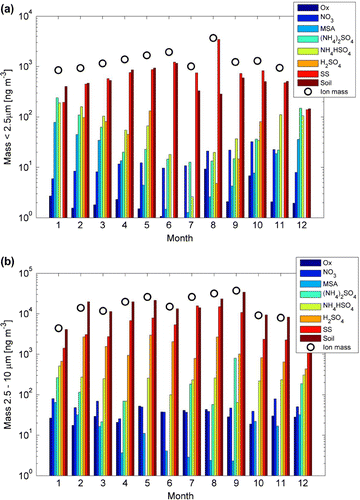
Figure 7. Average chemical composition of (a) fine mode (2.5
m) and (b) coarse mode (2.5–10
m) particles in different months. Colours from blue to red present the mass concentrations of oxalate, nitrate, methanesulphonic acid, ammonium sulphate, ammonium bisulphate, sulphuric acid, sea salt and soil. Black circles show the total mass calculated as the sum of chemical compounds.
Similar to some previous studies (Hara et al., Citation2008; Weller et al., Citation2013), wind speed was found to be connected with increasing EBC concentration. Indeed, at wind speeds 15 m s
(represent 16% of data) both the aerosol scattering and absorption were found to be strongly elevated (Fig. ). Interestingly, the increasing scattering and absorption values mostly compensated each other, leading to a very minor decrease in the SSA with wind speed. And at the lowest wind speeds (
10 m s
), the SSA actually increased with wind. The SAE decreased with wind speed, indicating that the observed increase in scatter and absorption was mostly due to coarse, than fine mode aerosols. In previous studies wind speed impact on aerosol absorption has mainly been explained by changing transport patterns (Hara et al., Citation2008; Weller et al., Citation2013), while in Marambio the simultaneous impact seen on scattering would suggest the influence of primary sources, such as sea spray or wind-blown dust particles. This will further be examined using the measured ionic compositions.
3.4. Chemical composition
Aerosol chemical composition can help in interpreting the measured particle optical properties and their variability. Based on 3-years of continuous weekly filter collections, we can conclude that sea salt particles constitute a major fraction of both fine (67%) and coarse (30%) mode aerosol mass in Marambio. As sea salt is a purely light scattering aerosol, it is likely to contribute most to the observed values. Sea salt concentration increased by a factor of 4–5 when wind speed increased to
12 m s
(Tables and ). In winter, the averaged wind speed was
12 m s
for 24% of the filters. While high winds were less frequent during summer, a lower wind speed limit of 10 m s
was used in the analysis to obtain the same statistical significance (24% of the summertime cases). Most likely due to this limitation, also the increase in sea salt concentration with wind speed during summer was less apparent in the results.
Other significant compounds which were present both in summer and winter were nss-fractions of sulphate and calcium. While the concentration of ammonium, which is the main neutralizing compound of sulphate, was relatively low, the aerosol sulphate was mainly in the form of sulphuric acid. Low ammonium and nitrate concentrations also suggest that the long-range transported aerosol fraction was minor. A major source of nss-sulphate in Antarctica is the oxidation of oceanic dimethyl sulphide (DMS). Another oxidation product of DMS is MSA, which was mainly present during summer due to its sole oxidation pathway via the OH-radical. The nss-sulphate to MSA ratio was around 0.25 in fine mode aerosol, but much lower (around 0.02) in coarse mode aerosol during summer (Tables and ). This also suggests that none marine sources contribute to coarse mode high sulphate loadings, such as the anthropogenic activities around the region of the Antarctic Peninsula or aerosol long-range transport and further resuspension of the aerosol as primary particles.
High fractions of nss-Ca both during summer and winter, as well as its increasing concentration during high wind speed points to a significant fraction of crustal mineral particles originating from Antarctic coastal soils or from further distances via long-range transport. Assuming that our calculation of nss-Ca concentration which is based on standard seawater calcium fraction is a valid approximation, the soil originated particles indeed seem to be present at approximately equal concentrations as sea salt in the fine aerosol mode, and at even higher concentrations in the coarse aerosol mode throughout the year (Fig. and Tables S2 and S3). Both sea spray and mineral dust aerosol concentrations show an increase with wind speed that here is approximately equal in magnitude, thereby explaining the observed increase of (Tables and , Fig. S3). Also in some previous studies Antarctic coastal soil minerals have been noted as a significant contributor to the total aerosol mass especially in the Peninsula region (Artaxo and Rabello, Citation1992; Pereira et al., Citation2004; Weller et al., Citation2008; DiasdaCunha et al., Citation2009; Gao et al., Citation2013; Xu and Gao, Citation2014). Which fraction is local and which originates from long-range transport however, is not clear. Pereira et al. (Citation2004) suggested that even 95% of the insoluble deposited particles collected from Lange Glacier actually originate from Chilean Patagonia. Based on our calculated back-trajectories, a minor fraction of the aerosol can be transported to Marambio from the southernmost parts of South-America. However, due to the prevalence of relatively short-lived coarse mode aerosol, we assume that most of the dust is from locally generated Antarctic origin. Also it should be noted that calcium is not the best marker for mineral dust due to its presence in the seawater. It has also been found to be significantly enriched in Antarctic coasts and seawater, which suggests additional anthropogenic sources of
are present (Préndez et al., Citation2009; Budhavant et al., Citation2015). Calcium enrichment has also been observed in the Arctic as suggested by very recent evidence by Salter et al. (Citation2016).
Based in Tables and and Fig. it is clear that most of the particle mass is in the coarse mode. This further points towards major primary particle sources, and explains the observed weak wavelength dependence of particle scattering. In winter (April to September) the average fine mode weighted mass was 4.7 2.2
g m
when wind speed was
12 m s
(24% of filters) and 2.8
1.9
g m
when wind speed was
12 m s
. The concentration of sea salt increased by a factor of 5 and the concentration of soil particles by a factor of 2.5 during higher wind speed (Fig. S3, a-b). The concentration of nss-sulphate increased on average by a factor of 10, suggesting a similar origin, likely re-suspension from soils and sea water as part of primary aerosol. Also concentrations of other measured ions increased slightly with wind speed. In summer (October to March) the average fine mode weighted mass was 5.0
4.9
g m
when wind speed was
10 m s
(24% of filters) and 2.1
1.1
g m
when wind speed was
10 m s
. The effect of wind speed on ion mass was less visible in summer due to the lower frequency of storms and the relatively long collection period of about a week (Fig. S3, c and d). However, the concentrations of sea salt and soil particles on average nearly doubled also in summer when winds were high. In contrast to the winter fine mode, no increase with wind speed was observed in nss-sulphate and MSA during summer. This is likely explained by secondary sources of these compounds via more efficient OH-radical oxidation in summer.
The coarse mode filters were not weighted but based on average chemical composition calculated from ions, the coarse mode mass concentration was much higher than that of fine mode (Table ) (Fig. S3, e–h). Based on calculated chemical mass balance of measured ions, the sum of compounds in fine mode was on average 1.48 3.39
g m
with a median of 0.97
g m
and in coarse mode 18.3
33.0
g m
with a median of 7.32
g m
. Similar to fine mode, most of the particle mass in coarse mode was composed of sea salt and soil particles, with some nss-sulphate and minor fractions of other compounds (Fig. ). Altogether, primary aerosol sources control the mass composition in Marambio at all times. Higher concentrations of MSA and fine mode nss-sulphate in summer however suggest that secondary oxidation via OH-radical contributes to formation of these compounds. Also ammonium concentration is increased during summer, suggesting additional sources, such as penguin colonies (Croft et al., Citation2016).
While the aerosol origin and chemical composition provide an explanation for the measured aerosol scattering and its variability, they are not quite able to explain the relatively constant SSA observed throughout the year. In most parts of the Antarctica the aerosol absorbing fraction is mainly composed of black ‘soot’ particles (Warren and Clarke, Citation1990; Fiebig et al., Citation2009). However, also mineral dust particles can be weakly absorbing. Absorption of pure mineral dust particles is about 100–1000 times less than that of EBC, and a SSA for dust aerosol at wavelengths 500 nm typically ranges from 0.98 to close to one (Linke et al., Citation2006; Moosmüller et al., Citation2012) but decreases drastically at smaller wavelengths. Absorption mainly depends on dust chemical composition and is highly dependent on soil iron content (Derimian et al., Citation2008; Moosmüller et al., Citation2012). Mineral dust particle morphology and size also have effects on their absorption efficiency. Mixed with EBC, aerosol absorption changes depending on all the above mentioned quantities, but mainly the size distribution of dust aerosol (Clarke et al., Citation2004; Derimian et al., Citation2008; Müller et al., Citation2009; Scarnato et al., Citation2015). Typically for larger dust particles mixed with EBC, the aerosol total SSA decreases. Thus, it is fairly complicated to distinguish the absorbing aerosol chemical composition and especially to separate the black carbon, or soot, fraction from other contributions to the absorbing aerosol. Additional absorbing compounds such as organic carbon are also likely present in the vicinity of the biologically active Southern Ocean (Asmi et al., Citation2010; Legrand et al., Citation2013).
Mie calculations can be used to give a rough estimate of the absorption by mineral dust particles. We used the Mie code by Barber and Hill (Citation1990) to calculate the aerosol extinction and scattering efficiencies. Based on our filter data, we could estimate that about half of the particles were sea salt, while the other half was mineral dust. The refractive index used for the equal volume mixed sea salt-mineral dust aerosol was , assuming that the imaginary part (k) of dust refractive index (
) at wavelength 635 nm was 0.001. This is an upper-limit estimate, based on Clarke et al. (Citation2004) and Müller et al. (Citation2009), while the exact soil dust composition here was unknown. Mie theory also assumes spherical particles which especially for mineral dust aerosol might induce some source of error. We calculated the scattering vs. extinction efficiency (i.e. SSA) for two particle mode mean diameters: 1 and 5
m. The SSA for the smaller particle mode (1
m) became 0.996, while the SSA for the larger particle mode (5
m) was 0.97. This suggests that coarse mode mineral dust can have a noticeable impact on aerosol light absorption. However to conclude this, we should better determine the dust aerosol elemental composition and size distribution at our site. Based on earlier measurements in the Antarctica we assume that also some soot is present. Without additional measurements on the wavelength dependence of absorption, particle size distribution, and mineral dust elemental composition, based on absorption measurements only it is therefore impossible to estimate the soot aerosol mass fraction quantitatively.
4. Summary and conclusions
Aerosol scattering and absorption coefficients were measured continuously at the Marambio station in the Antarctic Peninsula during three years (2013–2015). The average (median) scattering coefficients were Mm
,
Mm
, and
Mm
, and the absorption coefficient
was 0.11 (0.05) Mm
. Aerosol was highly scattering showing an average SSA at
nm of 0.96
0.10, with a median of 0.99. Scattering coefficients were in the same order of magnitude or slightly higher than those observed at the coastal Neumayer site (Weller and Lampert, Citation2008a), and showed a similar wintertime maximum mainly due to increased marine sea salt emissions. The wavelength dependence of scattering in Marambio however was much weaker than observed at the other stations, and the obtained average scattering SAE of 0.6
2.0 indicates dominance of coarse mode particles. SAE increased during summer indicating an additional contribution from secondary particle sources, which is supported by changes in particle chemical composition.
Equivalent black carbon (EBC) mass concentrations were measured by optical means. The measurements showed overall average and median EBC concentrations of 17.3 and 6.9 ng m. This is clearly higher than previously measured at the coastal Halley and Neumayer sites (Wolff and Cachier, Citation1998; Weller et al., Citation2013), and on the same order of magnitude as observed at the Ferraz station on the Antarctic Peninsula (Pereira et al., Citation2006) and at the eastern Antarctic coastal sites Syowa and Larsemann Hills (Hara et al., Citation2008; Chaubey et al., Citation2010), but lower than EBC measured at the Indian station Maitri (Chaubey et al., Citation2010). In contrast to most earlier studies, no clear seasonal cycle in EBC was found.
In agreement with previous studies (Hara et al., Citation2008; Weller et al., Citation2013), EBC concentration was shown to increase with wind speed. However, in Marambio this was largely attributed to the dominance of primary aerosol sources (sea salt and mineral dust), leading to an almost equal increase of both aerosol scattering and absorption, while in previous studies the intensified long-range transport during high winds was suggested as the main cause for the observed EBC increase. Based on back-trajectories a small fraction of measured air-masses also originated in South-America but their significance as a source of absorbing black carbon remained unclear.
The aerosol mass concentration measured in Marambio was relatively high compared to other sites in Antarctica. The average weighted fine mode (2.5
m) mass concentration was 2.1 and 2.8
g m
in summer and winter, respectively, but increased to 5.0 and 4.7
g m
during higher wind speeds. The sum of the analyzed fine mode chemical compounds was on average 1.48
3.49
g m
, indicating that a fraction (
40%) of particle mass was missing. While the filters were weighted at 50% RH, solely the particle hygroscopic growth with water might be enough to explain this difference. However, we may not exclude that the organic aerosol fraction which was not measured here but has been observed in previous Antarctic studies (Asmi et al., Citation2010), forms a part of the missing mass. The coarse mode mass concentration calculated as a sum of chemical compounds was on average 12-fold, and on median sevenfold, the fine mode mass concentration. Thus most of the particle mass were in the coarse mode, and the increasing primary emissions with wind were the main cause for the deviation of average and median mass concentration values. Also previous studies have reported relatively similar mass concentrations on the order of some
g m
measured at the Antarctic coastal sites (Artaxo and Rabello, Citation1992; Mazzera et al., Citation2001; Chaubey et al., Citation2011; Budhavant et al., Citation2015), yet not such a significant increase towards the coarse mode was observed before. All studies, however, are consistent with mass concentration seasonal cycle showing higher values in winter.
The aerosol in Marambio was mainly composed of sea salt, sulphate and crustal soil minerals. In summer, secondary sulphate and MSA were elevated in fine mode and the increasing ammonium concentration could neutralize most of the fine mode sulphate, opposite to winter. The chemical composition observed in Marambio is consistent with previous Antarctic observations, but the soil particle mass, which was calculated based on nss- fraction, was higher than reported at other coastal stations. Windblown dust from open soils on the Peninsula is suggested as the main source of these particles, based on the typical air mass back-trajectories circulating towards Marambio along the coastal Peninsula. A fraction of fine mode dust could also originate from South-America, as suggested in some previous studies, but it is unlikely to be the main source region while most of the coarse mode aerosol would deposit well before reaching Antarctica. In contrast, for smaller black carbon aerosols this could be a significant source region, which was however not evidenced by our back-trajectory analysis. Soil dust aerosol is also weakly absorbing, and at high quantities observed, it was shown to be able to explain a fraction of aerosol absorption in Marambio. This is in line with increasing absorption observed at wind speeds
15 m s
, caused by the wind-driven suspension of soil dust. Nevertheless, absorbing aerosol is most likely a mixture of dust minerals and soot, but additional measurements of soil mineral composition and wavelength dependence of absorption are needed to define their quantitative absorbing fractions.
Supplementary Material
Download PDF (860.4 KB)Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
Supplemental data for this article can be accessed here: https://doi.org/10.1080/16000889.2017.1414571.
Related Research Data
References
- AMAP. (2011). In: The Impact of Black Carbon on Arctic Climate (eds. Quinn, P.K., Stohl, A., Arneth, A., Berntsen, J.F., Christensen, J., Flanner, M., Kupiainen, K., Lihavainen, H., Shepherd, M., Shevchenko, V., Skov, H., and Vestreng, V. ) Oslo: Arctic Monitoring and Assessment Programme (AMAP). 72 pp.
- Artaxo, P. and Rabello, M. L. C. 1992. Trace elements and individual particle analysis of atmospheric aerosols from the Antarctic Peninsula. Tellus 44B, 318–334.
- Asmi, E., Frey, A., Virkkula, A., Ehn, M., Manninen, H. E. and co-authors. 2010. Hygroscopicity and chemical composition of Antarctic sub-micrometre aerosol particles and observations of new particle formation. Atmos. Chem. Phys. 10, 4253–4271. DOI:10.5194/acp-10-4253-2010.
- Atkinson, H. M., Huang, R.-J., Chance, R., Roscoe, H. K., Hughes, C. and co-authors. 2012. Iodine emissions from the sea ice of the Weddell Sea. Atmos. Chem. Phys. 12, 11229–11244. DOI:10.5194/acp-12-11229-2012.
- Barber, P. W. and Hill, S. C. 1990. Light Scattering by Particles: Computational Methods World Scientific, Singapore.
- Bisiaux, M. M., Edwards, R., McConnell, J. R., Curran, M. A. J., Van Ommen, T. D. and co-authors. 2012. Changes in black carbon deposition to Antarctica from two high-resolution ice core records, 1850–2000 AD. Atmos. Chem. Phys. 12, 4107–4115. DOI:10.5194/acp-12-4107-2012.
- Bodhaine, B. A., Deluisi, J. J. and Harris, J. M. 1986. Aerosol measurements at the South Pole. Tellus 38B, 223–235.
- Budhavant, K., Safi, P. D. and Rao, P. S. P. 2015. Sources and elemental composition of summer aerosols in the Larsemann Hills (Antarctica). Environ. Sci. Pollut. Res. 22, 2041–2050. DOI:10.1007/s11356-014-3452-0.
- Chaubey, J. P., Moorthy, K. K., Babu, S. S. and Nair, V. S. 2011. The optical and physical properties of atmospheric aerosols over the Indian Antarctic stations during southern hemispheric summer of the International Polar Year 2007–2008. Ann. Geophys. 29, 109–121. DOI:10.5194/angeo-29-109-2011.
- Chaubey, J. P., Moorthy, K. K., Babu, S. S., Nair, V. S. and Tiwari, A. 2010. Black carbon aerosols over coastal Antarctica and its scavenging by snow during the Southern Hemispheric summer. J. Geophys. Res. 115, D10210. DOI:10.1029/2009JD013381.
- Clarke, A. D., Shinozuka, Y., Kapustin, V. N., Howell, S., Huebert, B. and co-authors. 2004. Size distributions and mixtures of dust and black carbon aerosol in Asian outflow: physiochemistry and optical properties. J. Geophys. Res. 109, D15S09. DOI:10.1029/2003JD004378.
- Croft, B., Wentworth, G. R., Martin, R. V., Leaitch, W. R., Murphy, J. G. and co-authors. 2016. Contribution of Arctic seabird-colony ammonia to atmospheric particles and cloud-albedo radiative effect. Nat. Commun. 7, 13444. DOI:10.1038/ncomms13444.
- Derimian, Y., Karnieli, A., Kaufman, Y. J., Andreae, M. O., Andreae, T. W. and co-authors. 2008. The role of iron and black carbon in aerosol light absorption. Atmos. Chem. Phys. 8, 3623–3637. DOI:10.5194/acp-8-3623-2008.
- DiasdaCunha, K., Medeiros, G., Leal, M. A., Lima, C., Dalia, K. C. and co-authors. 2009. Aerosols in King George Island (Antarctic Peninsula) using PIXE and ALPHA spectrometry. 2009 International Nuclear Atlantic Conference -- INAC 2009. ISBN: 978-85-99141-03-8.
- Draxler, R. R. 1999. HYSPLIT4 user’s guide. NOAA Tech. Memo. ERL ARL-230 NOAA Air Resources Laboratory, Silver Spring, MD.
- Draxler, R. R. and Hess, G. D. 1997. Description of the HYSPLIT4 modeling system. In: NOAA Tech. Memo. ERL ARL-224 NOAA Air Resources Laboratory, Silver Spring, MD, p. 27.
- Draxler, R. R. and Hess, G. D. 1998. An overview of the HYSPLIT4 modeling system of trajectories, dispersion, and deposition. Aust. Meteor. Mag. 47, 295–308.
- Fiebig, M., Hirdman, D., Lunder, C. R., Ogren, J. A., Solberg, S. and co-authors. 2014. Annual cycle of Antarctic baseline aerosol: controlled by photooxidation-limited aerosol formation. Atmos. Chem. Phys. 14, 3083–3093. DOI:10.5194/acp-14-3083-2014.
- Fiebig, M., Lunder, C. R. and Stohl, A. 2009. Tracing biomass burning aerosol from South America to Troll Research Station, Antarctica. Geophys. Res. Lett. 36, L14815. DOI:10.1029/2009GL038531.
- Gao, Y., Xu, G., Zhan, J., Zhang, J., Li, W. and co-authors. 2013. Spatial and particle size distributions of atmospheric dissolvable iron in aerosols and its input to the Southern Ocean and coastal East Antarctica. J. Geophys. Res. Atmos. 118, 12634–12648. DOI:10.1002/2013JD020367.
- Giordano, M. R., Kalnajs, L. E., Avery, A., Goetz, J. D., Davis, S. M. and co-authors. 2017. A missing source of aerosols in Antarctica - beyond long-range transport, phytoplankton, and photochemistry. Atmos. Chem. Phys. 17, 1–20. DOI:10.5194/acp-17-1-2017.
- Graf, H.-F., Shirsat, S. V., Oppenheimer, C., Jarvis, M. J., Podzun, R. and co-authors. 2010. Continental scale Antarctic deposition of sulphur and black carbon from anthropogenic and volcanic sources. Atmos. Chem. Phys. 10, 2457–2465. DOI:10.5194/acp-10-2457-2010.
- Hansen, A. D. A., Bodhaine, B. A., Dutton, E. G. and Schnell, R. C. 1988. Aerosol black carbon measurements at the South Pole: initial results, 1986–1987. Geophys. Res. Lett. 15, 1193–1196.
- Hansen, D. A. A., Lowenthal, D. H., Chow, J. C. and Watson, J. G. 2001. Black Carbon Aerosol at McMurdo Station, Antarctica. J Air & Waste Manage. Assoc. 51(4), 593–600. DOI:10.1080/10473289.2001.10464283.
- Hansen, J. and Nazarenko, L. 2004. Proc. Natl. Acad. Sci. 101, 423–428. DOI:10.1073/pnas.2237157100.
- Hara, K., Osada, K., Yabuki, M., Hayashi, M., Yamanouchi, T. and co-authors. 2008. Measurement of black carbon at Syowa station, Antarctica: seasonal variation, transport processes and pathways. Atmos. Chem. Phys. Discuss. 8, 9883–9929. DOI:10.5194/acpd-8-9883-2008.
- Hu, Q. H., Xie, Z. Q., Wang, X. M., Kang, H. and Zhang, P. 2013. Levoglucosan indicates high levels of biomass burning aerosols over oceans from the Arctic to Antarctic. Sci. Rep. 3, 3119. DOI:10.1038/srep03119.
- Hyvärinen, A.-P., Vakkari, V., Laakso, L., Hooda, R. K., Sharma, V. P. and co-authors. 2013. Correction for a measurement artifact of the Multi-Angle Absorption Photometer (MAAP) at high black carbon mass concentration levels. Atmos. Meas. Tech. 6, 81–90. DOI:10.5194/amt-6-81-2013.
- Ito, T. 1989. Antarctic submicron aerosols and long-range transport of pollutants. Ambio 18(1), 34–41.
- Jourdain, B., Preunkert, S., Cerri, O., Castebrunet, H., Udisti, R. and Legrand, M. 2008. Year-round record of size-segregated aerosol composition in central Antarctica (Concordia station): Implications for the degree of fractionation of sea-salt particles. J. Geophys. Res. 113, D14308. DOI:10.1029/2007JD009584.
- Karhu, J. A., Taalas, P., Damski, J., Kaurola, J., Ginzburg, M., Villanueva, C. A., Piacentini, E. and Garcia, M. 2003. Vertical distribution of ozone at Marambio, Antarctic Peninsula, during 1987–1999. J. Geophys. Res. 108(D17), 4545. DOI:10.1029/2003JD001435.
- Kaspari, S., Dixon, D. A., Sneed, S. B. and Handley, M. J. 2005. Sources and transport pathways of marine aerosol species into West Antarctica. Ann. Glaciol. 41(1), 1–9. DOI:10.3189/172756405781813221.
- Kyrö, E.-M., Kerminen, V.-M., Virkkula, A., Dal Maso, M. and Parshintsev, J., and co-authors. 2013. Antarctic new particle formation from continental biogenic precursors. Atmos. Chem. Phys. 13, 3527–3546. DOI:10.5194/acp-13-3527-2013.
- Legrand, M., Preunkert, S., Jourdain, B., Guilhermet, J., Fa, X. and co-authors. 2013. Water-soluble organic carbon in snow and ice deposited at Alpine, Greenland, and Antarctic sites: a critical review of available data and their atmospheric relevance. Clim. Past 9, 2195–2211. DOI:10.5194/cp-9-2195-2013.
- Linke, C., Möhler, O., Veres, A., Mohácsi, A., Bozóki, Z. and co-authors. 2006. Optical properties and mineralogical composition of different Saharan mineral dust samples: a laboratory study. Atmos. Chem. Phys. 6, 3315–3323. DOI:10.5194/acp-6-3315-2006.
- Loo, B. W. and Cork, C. P. 1988. Development of high efficiency virtual impactors. Aerosol. Sci. Technol. 9, 167–176.
- Mattsson, R., Paatero, J. and Hatakka, J. 1996. Automatic Alpha/Beta Analyser for Air Filter Samples - Absolute Determination of Radon Progeny by Pseudo-coincidence Techniques. Radiat. Prot. Dosim. 63, 133–139.
- Mazzera, D. M., Lowenthal, D. H., Chow, J. C., Watson, J. G. and Grubísíc, V. 2001. PM10 measurements at McMurdo Station, Antarctica. Atmos. Env. 35, 1891–1902.
- Meinander, O., Aarva, A., Poikonen, A., Kontu, A., Suokanerva, H. and co-authors. 2016. Bipolar long-term high temporal resolution broadband measurement system for incoming and outgoing solar UV radiation, and snow UV albedo, at Sodankyl (67°N) and Marambio (64°S). Geosci. Instrum. Method. Data Syst. Discuss.. DOI:10.5194/gi-2015-31 in review.
- Moosmüller, H., Engelbrecht, J. P., Skiba, M., Frey, G., Chakrabarty, R. K. and co-authors. 2012. Single scattering albedo of fine mineral dust aerosols controlled by iron concentration. J. Geophys. Res. 117, D11210. DOI:10.1029/2011JD016909.
- Müller, T., Henzing, J. S., de Leeuw, G., Wiedensohler, A., Alastuey, A. and co-authors. 2011. Characterization and intercomparison of aerosol absorption photometers: result of two intercomparison workshops. Atmos. Meas. Tech. 4, 245–268. DOI:10.5194/amt-4-245-2011.
- Müller, T., Laborde, M., Kassell, G. and Wiedensohler, A. 2011. Design and performance of a three-wavelength LED-based total scatter and backscatter integrating nephelometer. Atmos. Meas. Tech. 4, 1291–1303. DOI:10.5194/amt-4-1291-2011.
- Müller, T., Schladitz, A., Massling, A., Kaaden, N., Kandler, K. and co-authors. 2009. Spectral absorption coefficients and imaginary parts of refractive indices of Saharan dust during SAMUM-1. Tellus B 61, 79–95. DOI:10.3402/tellusb.v61i1.16816.
- Murphy, D., Anderson, J., Quinn, P., McInnes, L., Brechtel, F. and co-authors. 1998. Influence of sea-salt on aerosol radiative properties in the Southern Ocean marine boundary layer. Nature 392, 62–65. DOI:10.1038/32138.
- Pereira, E. B., Evangelista, H., Pereira, K. C. D., Cavalcanti, I. F. A. and Setzer, A. W. 2006. Apportionment of black carbon in the South Shetland Islands, Antarctic Peninsula. J. Geophys. Res. 111, D03303. DOI:10.1029/2005JD006086.
- Pereira, K. C. D., Evangelista, H., Pereira, E. B., Simões, J. C., Johnson, E. and co-authors. 2004. Transport of crustal microparticles from Chilean Patagonia to the Antarctic Peninsula by SEM-EDS analysis. Tellus 56B, 262–275.
- Préndez, M., Wachter, J., Vega, C., Flocchini, R. G., Wakayabashi, P. and co-authors. 2009. PM2.5 aerosols collected in the Antarctic Peninsula with a solar powered sampler during austral summer periods. Atmos. Environ. 43, 5575–5578. DOI:10.1016/j.atmosenv.2009.07.030.
- Salter, M., Hamacher-Barth, E., Leck, C., Werner, J., Johnson, C. and co-authors. 2016. Calcium Enrichment in Sea Spray Aerosol Particles. Geophys. Res. Lett. 43, 8277–8285. DOI:10.1002/2016GL070275.
- Scarnato, B. V., China, S., Nielsen, K. and Mazzoleni, C. 2015. Perturbations of the optical properties of mineral dust particles by mixing with black carbon: a numerical simulation study. Atmos. Chem. Phys. 15, 6913–6928. DOI:10.5194/acp-15-6913-2015.
- Shaw, G. E. 1988. Antarctic aerosols: a review. Rev. Geophys. 26(1), 89–112.
- Stohl, A. and Sodemann, H. 2010. Characteristics of atmospheric transport into the Antarctic troposphere. J. Geophys. Res. 115, D02305. DOI:10.1029/2009JD012536.
- Tang, I. N. 1996. Chemical and size effects of hygroscopic aerosols on light scattering coefficients. J. Geophys. Res. 101(D14), 19245–19250.
- Tang, I. N., Tridico, A. and Fung, K. 1997. Thermodynamic and optical properties of sea salt aerosols. J. Geophys. Res. 102(D19), 23269–23275.
- Teinilä, K., Frey, A., Hillamo, R., Tülp, H. C. and Weller, R. 2014. A study of the sea-salt chemistry using size-segregated aerosol measurements at coastal Antarctic station Neumayer. Atmos. Env. 96, 11–19. DOI:10.1016/j.atmosenv.2014.07.025.
- Teinilä, K., Kerminen, V.-M. and Hillamo, R. 2000. A study of size-segregated aerosol chemistry in the Antarctic atmosphere. J. Geophys. Res. 105(D3), 3893–3904.
- Turner, J., Lu, H., White, I., King, J. C., Phillips, T. and co-authors. 2016. Absence of 21st century warming on Antarctic Peninsula consistent with natural variability. Nature 535, 411–415. DOI:10.1038/nature18645.
- Vaughan, D. G. 2006. Recent trends in melting conditions on the Antarctic Peninsula and their implications for ice-sheet mass balance. Arct. Antarct. Alp. Res. 38, 147–152.
- Virkkula, A., Teinilä, K., Hillamo, R., Kerminen, V.-M., Saarikoski, S. and co-authors. 2006. Chemical composition of boundary layer aerosol over the Atlantic Ocean and at an Antarctic site. Atmos. Chem. Phys. 6, 3407–3421. DOI:10.5194/acp-6-3407-2006.
- Wagenbach, D., Ducroz, F., Mulvaney, R., Keck, L., Minikin, A. and co-authors. 1998. Sea salt aerosol in coastal Antarctic regions. J. Geophys. Res. 103(D9), 10961–10974.
- Warren, S. G. and Clarke, A. D. 1990. Soot in the atmosphere and snow surface of Antarctica. J. Geophys. Res. 95(D2), 1811–1816.
- Weller, R. and Lampert, A. 2008a. Optical properties and sulfate scattering efficiency of boundary layer aerosol at coastal Neumayer Station, Antarctica. J. Geophys. Res. 113, D16208. DOI:10.1029/2008JD009962.
- Weller, R., Minikin, A., Petzold, A., Wagenbach, D. and König-Langlo, G. 2013. Characterization of long-term and seasonal variations of black carbon (BC) concentrations at Neumayer, Antarctica. Atmos. Chem. Phys. 13, 1579–1590. DOI:10.5194/acp-13-1579-2013.
- Weller, R., Schmidt, K., Teinilä, K. and Hillamo, R. 2015. Natural new particle formation at the coastal Antarctic site Neumayer. Atmos. Chem. Phys. 15, 11399–11410. DOI:10.5194/acp-15-11399-2015.
- Weller, R., Wöltjen, J., Piel, C., Resenberg, R., Wagenbach, D. and co-authors. 2008. Seasonal variability of crustal and marine trace elements in the aerosol at Neumayer station, Antarctica. Tellus 60B, 742–752. DOI:10.1111/j.1600-0889.2008.00372.x.
- Wolff, E. W. and Cachier, H. 1998. Concentrations and seasonal cycle of black carbon in aerosol at a coastal Antarctic station. J. Geophys. Res. 103(D9), 11033–11041.
- Wouters, B., Martin-Español, A., Helm, V., Flament, T., van Wessem, J. M. and co-authors. 2015. Dynamic thinning of glaciers on the Southern Antarctic Peninsula. Science 348(6237), 899–903. DOI:10.1126/science.aaa5727.
- Xu, G. and Gao, Y. 2014. Atmospheric trace elements in aerosols observed over the Southern Ocean and coastal East Antarctica. Polar Res. 33, 23973. DOI:10.3402/polar.v33.23973.
- Zieger, P., Fierz-Schmidhauser, R., Gysel, M., Strm, J., Henne, S., and co-authors. 2010. Effects of relative humidity on aerosol light scattering in the Arctic. Atmos. Chem. Phys. 10, 3875–3890. DOI:10.5194/acp-10-3875-2010.