
ABSTRACT
Objectives and importance: Chronic neutrophilic leukemia (CNL) is a distinct myeloproliferative neoplasm with a high prevalence (>80%) of mutations in the colony-stimulating factor 3 receptor (CSF3R); these mutations activate the receptor, leading to the proliferation of neutrophils that are a hallmark of CNL.
Clinical presentation: We present a male patient who presented peripheral blood leukocytosis. On the basis of his morphological appearances and molecular findings he was determined to have a diagnosis of chronic neutrophilic leukemia. At a follow-up at 7 months, in addition to the CSF3R c.2373G > A (p.W791*) truncated mutation, another CSF3R mutation appeared as c.1853C > T(p.T618I).
Discussion and conclusion: We present the first patient with a diagnosis of chronic neutrophilic leukemia with a c.2373G > A (p.W791*) truncated mutation of CSF3R. These findings elucidate a novel paradigm of CNL pathogenesis and explain how mutations drive the development of the disease. The order of acquisition of CSF3R mutations relative to mutations in epigenetic modifiers and the spliceosome have been determined only in isolated case reports; thus, further work is needed to understand the impact of mutation chronology on the clonal evolution and progression of CNL.
Introduction
Chronic neutrophilic leukemia (CNL) is a distinct myeloproliferative neoplasm with a high prevalence (>80%) of mutations in the colony-stimulating factor 3 receptor (CSF3R); these mutations activate the receptor, leading to the proliferation of neutrophils that are a hallmark of CNL. The WHO classification was updated in 2016 to activating mutations of CSF3R, such as CSF3R T618I, as a diagnostic tool. Two classes of mutations in CSF3R were identified: membrane-proximal mutations located in exon 14 and truncation mutations located in exon 17 [Citation1]. Both types of mutations were shown to possess in vitro transforming capacity, which is mediated via different downstream signaling pathways. Here, we describe the disease pathogenesis of an index CNL patient during a follow-up by identifying the clonal evolution.
Case presentation
A 66-year-old man was admitted to the Department of Hematology of the First People’s Hospital of Chuzhou with weight loss and fatigue. His past health history was unremarkable. At diagnosis, the patient presented peripheral blood leukocytosis (77.66×10^9/L), which were primarily composed of mature neutrophils (myelocytes 4%, metamyelocytes 4.5%, band neutrophils 30%, segmented neutrophils 57%, lymphocyte 3.5% and monocyte 1%). The patient showed moderate anemia (Hb 82 g/L) with a normal platelet count (150×10^9/L). Mild splenomegaly was present (below the costal margin of 5 cm). Bone marrow evaluation showed a markedly hypercellular with 83.8% myeloid cells (1% myeloblasts, 1.4% promyelocyte, 20% myelocyte, 24.8% metamyelocyte, 35.2% band and segmented neutrophils, 1% eosinophils, 0.4% basophil and 15.8% erythroid cells). No dysplastic features were observed in the myeloid lineages. Cytogenetic analyses revealed a normal karyotype with 20 metaphases. Molecular biology studies were negative for the JAK2, CALR and c-MPL genes and the BCR-ABL1 fusion gene, and the rearrangements of the PDGFRA, PDGFRB and FGFR1 genes were also negative. Next generation sequencing (NGS) was performed as previously described [Citation2]. The panel design was based on the Catalogue of Somatic Mutations in Cancer (COSMIC) database and literature searches, and included seven genes important in MDS/MPN. NGS revealed the presence of the SRSF2 (c.284C > A(p.P95H)) and TET2 (c.2293–2300del(p.S765fs*1)) mutations. CSF3R mutational analysis of the 14 and 17 exomes was performed by polymerase chain reaction (PCR) amplification followed by Sanger sequencing. A c.2373G > A (p.W791*) truncated mutation of CSF3R was detected ((a)). The patient was started on hydroxyurea and scheduled for a follow-up.
Figure 1. Detection of the W791* and T618I mutations in colony stimulating factor 3 receptor (CSF3R) during follow-up. (a) Sanger sequencing of exon 17 of CSF3R detected a base-pair mutation (G to A) at nucleotide 2373(p.W791*). (b) Sanger sequencing of exon 14 of CSF3R detected a base-pair mutation (C to T) at nucleotide 1853(p.T618I).
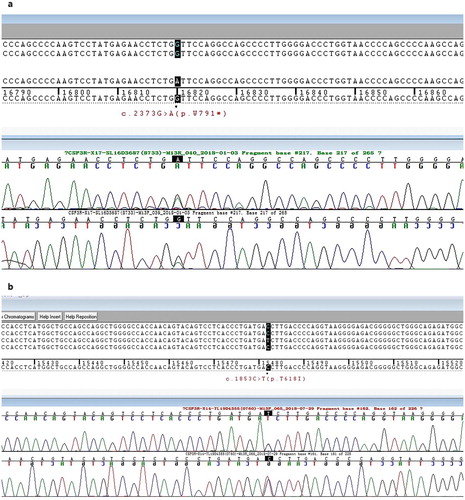
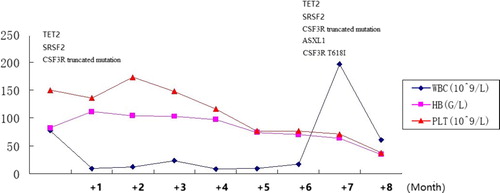
Chronologic overview of the clinical course of the CNL patient was shown in . At a follow-up at 7 months, the peripheral blood differentiation showed 2% myeloblasts with massive splenomegaly (below costal margin 20 cm). A second bone marrow aspirate was still hypercellular, with granulocytic hyperplasia with less than 10% immature granulocytes and no increase in the myeloblasts. In addition to the CSF3R c.2373G > A (p.W791*) truncated mutation, another CSF3R mutation appeared as c.1853C > T(p.T618I) ((b)). NGS also showed that the ASXL1 mutation (c.1934dupG (p.646fs)) occurred in addition to SRSF2 and TET2 mutations. The patient remained on hydroxyurea for economic reasons.
Figure 2. Chronologic overview of the clinical course of a CNL patient.
Discussion and conclusions
The current study, first, confirms the observations by Maxon et al. [Citation1] regarding the association between CNL and CSF3R mutations. Second, it presents a complete picture of the mutational profiling of the pathogenesis of CNL, which is certainly more complex than expected from previous reports. Approximately 75% of patients with CNL who have CSF3R mutations exhibit only membrane-proximal mutations, whereas the other 25% of patients harbor both membrane-proximal and truncating-compound mutations [Citation3]. The scarcity of truncation mutations alone in CNL raises the possibility that truncation mutations alone are insufficient to drive the malignancy and that cooperating mutations in CSF3R or other genes may be required [Citation4]. The CSF3R truncation mutation is not sufficient for leukemia but represents an early event in leukemogenesis. Truncation mutations alone do not cause leukemia in mouse models [Citation5], but they can accelerate the development of disease in the presence of another genetic event driving leukemia [Citation6]. Consistent with the in vivo data, the CSF3R truncation mutations display slower cell transformation kinetics than the T618I mutation in cellular assays and require ligands for the activation of downstream signaling pathways [Citation1,Citation7], which reduces the differentiative capacity of the receptor while continuing to allow for cellular proliferation. In our case, truncation mutations in the SRSF2 and TET2 genes are sufficient to drive leukemogenesis in the CNL, and CSF3R membrane-proximal mutations occur as a secondary event within the context of clonal hematopoiesis. The process of clonal hematopoiesis is originally associated with somatically acquired variants in genes that regulate epigenetic (TET2) and splicing (SRSF2) processes. Acquisition of a CSF3R signaling mutation gives the clonal hematopoietic population a neutrophil lineage bias. Because patients with CNL who have only truncation mutations in CSF3R are apparently rare, it seems likely that the clone evolves to acquire additional mutations in epigenetic and splicing genes, which amplify the disease process. Interestingly, concomitant SRSF2 and TET2 mutations were not observed in any other CNL cases, though this was not statistically significant [Citation8]. These mutations may provide a backdrop for a later-occurring CSF3R mutation, which, in turn, imparts a neutrophilic phenotype resulting in CNL.
SRSF2 is a member of the serine/arginine (SR) family of proteins that is characterized by 1 or 2 RNA recognition motifs (RRMs) and a serine/arginine-rich (SR) domain. RNA sequencing of the Srsf2-mutated hematopoietic stem and progenitor cells (HSPCs) revealed a number of abnormal splicing events and differentially expressed genes, such as Csf3r, Fyn, Gnas, Nsd1, Hnrnpa2b1, and Trp53bp1, which include several potential targets implicated in the pathogenesis of hematopoietic malignancies [Citation9].
Similar to mutations in other myeloid malignancies, ASXL1 mutation in the setting of CNL was shown to confer poor prognosis [Citation10]. In cellular and animal models, ASXL1 mutations have been shown to promote myeloid transformation through loss of PRC2-mediated gene repression [Citation11]. Evolution of CNL to CMML was observed in patients harboring ASXL1 mutations suggesting a pathogenic role for ASXL1 mutations in transformation to CMML [Citation10]. ASXL1 was presented at the time of progression of CNL (but not at diagnoses) and it also may contribute to the accelerate phase of the disease. These data indicate that the CNL genome had a combination of a few mutations with a pattern of cooperation with a strong biological relationship among genes and categories. The cooperative role of these and other spliceosome mutations in the pathogenesis of CNL, as well as their prognostic implications, remain to be determined.
Specifically, the JAK-STAT (activated by membrane-proximal mutations) and the SRC tyrosine kinase (activated by truncation mutations) pathways were involved, resulting in differential sensitivity of these receptors to kinase inhibitors such as ruxolitinib and dasatinib, respectively [Citation8]. BaF3 cells expressing CSF3R compound mutations constituting both membrane-proximal and truncation mutations in vitro were resistant to both JAK-STAT and SRC inhibitors [Citation12]. Enhanced MAPK signaling is crucial to leukemogenesis by CSF3R proximal and compound mutants. Therapeutic targeting of MEK1/2 by trametinib alone suppressed leukemia development in mice expressing either proximal or compound mutations [Citation12]. However, trametinib had less inhibitory effects than bosutinib in cell viability assays. Mechanistically, bosutinib effectively decreased the activity of the Src/Abl and PI3 K/AKT/mTOR, MAPK/ERK, and JAK/STAT3 signaling pathways. Our study provides a proof-of-concept for the therapeutic of bosutinib and establishes the rationale for the clinical evaluation of this concept.
These findings elucidate a novel paradigm of CNL pathogenesis and explain how mutations drive the development of the disease. The order of acquisition of CSF3R mutations relative to mutations in epigenetic modifiers and the spliceosome have been determined only in isolated case reports; thus, further work is needed to understand the impact of mutation chronology on the clonal evolution and progression of CNL.
Ethics approval and consent to participate
This case study was reviewed by Ethics Committee of The First People’s Hospital of Chuzhou City and determined that as it is based on information collected purely for clinical purposes, it is low risk and that formal ethical review is not required.
Acknowledgements
Not applicable.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. doi: 10.1056/NEJMoa1214514
- Reiman A, Kikuchi H, Scocchia D, et al. Validation of an NGS mutation detection panel for melanoma. BMC Cancer. 2017;17(1):150. doi: 10.1186/s12885-017-3149-0
- Gotlib J, Maxson JE, George TI, et al. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–1711. doi: 10.1182/blood-2013-05-500959
- Maxson JE, Tyner JW. Genomics of chronic neutrophilic leukemia. Blood. 2017;129(6):715–722. doi: 10.1182/blood-2016-10-695981
- Hermans MH, Ward AC, Antonissen C, et al. Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood. 1998;92(1):32–39.
- Kunter G, Woloszynek JR, Link DC. A truncation mutant of Csf3r cooperates with PML-RARα to induce acute myeloid leukemia in mice. Exp Hematol. 2011;39(12):1136–1143. doi: 10.1016/j.exphem.2011.08.013
- van de Geijn GJ, Gits J, Aarts LH, et al. G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood. 2004;104(3):667–674. doi: 10.1182/blood-2003-08-2913
- Meggendorfer M, Haferlach T, Alpermann T, et al. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica. 2014 Dec;99(12):e244–e246. doi: 10.3324/haematol.2014.113159
- Kon A, Yamazaki S, Nannya Y, et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood. 2018;131(6):621–635. doi: 10.1182/blood-2017-01-762393
- Elliott MA, Pardanani A, Hanson CA, et al. ASXL1 mutations are frequent and prognostically detrimental in CSF3R-mutated chronic neutrophilic leukemia. Am J Hematol. 2015;90(7):653–656. doi: 10.1002/ajh.24031
- Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–193. doi: 10.1016/j.ccr.2012.06.032
- Rohrabaugh S, Kesarwani M, Kincaid Z, et al. Enhanced MAPK signaling is essential for CSF3R-induced leukemia. Leukemia. 2017;31(8):1770–1778. doi: 10.1038/leu.2016.376