
ABSTRACT
Aplastic anemia (AA) is a rare and life-threatening bone marrow failure (BMF) that results in peripheral blood cytopenia and reduced bone marrow hematopoietic cell proliferation. The symptoms are similar to myelofibrosis, myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) making diagnosis of AA complicated. The pathogenesis of AA is complex and its mechanism needs to be deciphered on an individualized basis. This review summarizes several contributions made in trying to understand AA pathogenesis in recent years which may be helpful for the development of personalized therapies for AA.
1. Introduction
Aplastic anemia (AA) is a rare, life-threatening and heterogeneous disorder of the blood. It results in peripheral cytopenia with trilineage bone marrow (BM) aplasia. Anemia, bleeding, infection and several other clinical symptoms are usually the first presentations of AA. It may occur at any age, however young individuals (age 10–25 years) and the elderly (>60 years) are the most prone. No significant differences in gender have been noted [Citation1]. AA incidence in the United States and Europe is below 2.5/million, while the incidence of AA in Asia is 2–3 times higher [Citation2,Citation3]. However, the incidence rates of AA in Asia differ among the various countries, with rates of 7.4/million in China, 3.7–5.0/million in Thailand, and 4.8/million in Malaysia. Environmental factors, such as drugs, toxins and chemicals may influence the incidence of AA [Citation4].
AA can be divided into congenital and acquired. The inherited form is rare and mainly include Fanconi Anemia (FA), Congenital Keratosis (DKC), Congenital Pure Red Cell Aplasia(DBA) and Shwachman-Diamond Syndrome(SDS). Hematopoietic stem cell transplantation (HSCT) and anti-thymocyte globulin (ATG)-based immunosuppressive therapy (IST) have been the major treatment strategies for AA. However, the mechanism of AA is very complicated and has a high relapse rate with the secondary clonal disease. We reviewed the progress made in understanding the pathogenesis of AA in recent years so as to guide more effective clinical treatment strategies ().
Table 1. Summary of aplastic anemia pathogenesis.
2. Deficiencies in hematopoietic stem and progenitor cells (HSPCs)
Hematopoietic stem cells (HSCs) have the ability to self-renew and are pluripotent to differentiate into several hematopoietic lineages. They play an important role in the maintenance and regeneration of the hematopoietic system [Citation5]. Activated HSCs are responsible for the routine maintenance of hematopoiesis and tissue homeostasis. Quiescent subsets that form a stem cell reservoir could be activated after tissue damage to restore the normal stem cell pool and hematopoietic function [Citation6]. Accumulation of DNA damage in HSPCs during their lifespan is a factor responsible for the aging and degeneration of the hematopoietic system and may contribute to transformation and cancer development [Citation7]. It is hypothesized that AA is characterized by a loss or dysfunction of HSPCs. It involves both the quantitative loss of stem cell numbers and the qualitative abnormalities in stem cell function [Citation8,Citation9]. This was demonstrated by Maciejewski et.al where they showed a decrease in number and function of HSPCs using long-term culture initiating cell (LTC-IC) assays [Citation8]. External factors such as viruses, radiation and chemotherapeutic drugs affect HSC homeostasis, differentiation and self-renewal, making individuals vulnerable to AA [Citation10]. The three lineages that are derived from hematopoietic cells are significantly reduced in AA patients, while non-hematopoietic cells and adipocytes increase in proliferation. In addition, increased apoptosis of bone marrow progenitor cells (lin-c-kit+sca-1+CD34+) was observed in AA patients and maybe due to stem cell deficiencies [Citation11]. Maciejewski et.al showed that functional expression of Fas antigen on CD34+ cells was increased in AA patients compared to healthy individuals [Citation12]. It has been demonstrated that Fas-mediated apoptosis of CD34+ progenitor cells leads to HSC depletion [Citation13,Citation14]. Fas binds to FasL and is a member of the tumor necrosis factor receptor/neural growth factor receptor superfamily. Under physiological conditions, Fas is expressed on several cell surfaces, including activated T cells, B cells, monocytes and granulocytes to regulate proliferation and/or clearance [Citation12]. Timeus F et al. demonstrated that CD34+ cells in the peripheral blood of AA patients were lower and had higher rates of apoptosis compared to healthy individuals [Citation15]. Z.H.Shao et.al using flow cytometry examined 15 newly diagnosed SAA patients (9 males and 6 females). They found an increased level of apoptosis in bone marrow hematopoietic cells in SAA patients and concluded apoptosis was induced by the recognition of Fas expression antigen by FasL expressing cytotoxic T lymphocytes (CTL) [Citation12]. The above data suggests the involvement of Fas/FasL in the apoptosis of HSC and demonstrates a possible mechanism for dysfunction of bone marrow hematopoietic cells in SAA patients.
3. Abnormal bone marrow microenvironment
Another pathogenetic mechanism for AA may involve abnormal bone marrow microenvironment. Endosteal, vascular and perivascular cells make up the bone marrow microenvironment and play a significant role together with HSCs in hematopoiesis [Citation16]. Endosteum niche cells provide a quiescent HSC microenvironment by secreting regulatory molecules and cytokines [Citation17–19], while the vascular niche regulates the proliferation, differentiation and mobilization of HSCs [Citation20]. Liangliang Wu et.al analyzed the cellular components of the bone marrow microenvironment using in situ immunohistochemical staining. They found that AA patients had fewer endosteal, vascular and perivascular cells compared to healthy controls. This suggested AA was associated with impaired niches [Citation21]. In fact, little was known regarding niche cells until recently. Niche cells make up a small subset of non-hematopoietic bone progenitor cells called mesenchymal stem cells (MSCs). MSCs can differentiate into osteoblasts, chondrocytes and adipocytes [Citation22], and secrete a number of cytokines and growth factors that affect hematopoietic function through direct and paracrine mechanisms [Citation23]. MSCs in the bone marrow secrete interleukin (IL)-6, IL-11, IL-12 and flt-3 ligands, which affect the proliferation, differentiation and self-renewal of HSCs. They also secrete chemokine (CXCL)-12 which regulates the adhesion, expansion, migration and homing of HSCs, which in turn secrete several soluble mediators such as intercellular adhesion molecule-1 (ICAM-1) that interacts with T cells to regulate the immune response [Citation24,Citation25]. Compared to MSCs from healthy individuals, BM-MSCs from AA patients had reduced proliferation and were deficient in immune suppression of mixed lymphocyte reaction (MLR) and IFN-γ release. BM-MSCs from AA patients had the tendency to differentiate into adipocytes and had reduced expression of osteonectin [Citation26]. Unlike osteoblasts, adipocytes affect the proliferation and renewal of HSC [Citation27], resulting in bone marrow failure and hematopoietic cell loss. In view of the inhibitory effect of MSCs on the proliferation and cytotoxicity of immune cells, several clinical studies have demonstrated that co-transplantation of MSCs with allogeneic HSCs had tremendous improvement in hematopoietic function in AA patients (information on these studies were gathered from the National Institutes of Health (NIH) clinical trial database) [Citation25,Citation28]. However previous though was that the MSC phenotype and differentiation in the bone marrow of AA patients were normal and had immunomodulatory function [Citation29]. The hematopoietic microenvironment is complex and needs to be further investigated to understand the pathogenesis of AA.
4. Immune dysfunction
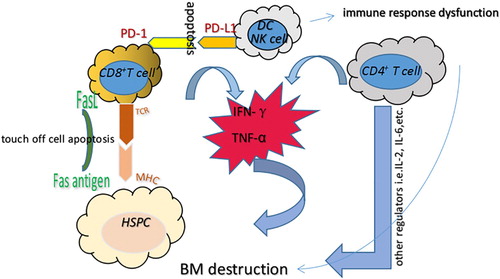
Recent clinical studies have suggested that AA is an autoimmune and bone marrow destructive disease that is mediated by abnormally activated T lymphocytes and their secreted lymphokines (). Neal s. Young et al. successfully constructed an immune-mediated bone marrow failure model by infusing 4–10 × 106 allogeneic lymph node (LN) cells into C57BL/6 (B6) mice that had undergone total-body irradiation (TBI) with 6.5 G [Citation30]. These mice had significantly fewer blood cells and severe bone marrow dysplasia compared to non-infused mice. The administration of immunosuppressants against human thymoglobulin (ATG) and cyclosporine (CSA) had a beneficial response rate of approximately 60–70%, with overall survival rates of 60–90% [Citation31–33]. This suggests that AA pathogenesis was associated with immune-mediated hematopoietic depletion. However, approximately 30–40% of AA patients after IST will relapse, suggesting that IST is not a cure.
Figure 1 . Immune-mediated mechanism related to AA pathogenesis. AA is thought to be mediated by abnormally activated T lymphocytes and their secreted lymphokines, which subsequently result in HSC dysfunction and BM destruction. Overproduction of pro-inflammatory cytokines, including IFN-γ, TNF-α and other regulators, inhibits the hematopoietic system and leads to cell apoptosis through the Fas/FasL signaling pathway. In addition, IFN-γ could induce PD-L1 expression on T cells, NK cells and dendritic cells, which then binds to PD-1 to induce apoptosis and reduce immune tolerance. DC: dendritic cell; HSC: hematopoietic stem cell; IFN-γ: interferon-γ; TNF-α: tumor necrosis factor-α.
4.1. Dendritic cells (DCs)
DCs are antigen presenting cells (APC) and were discovered by the Canadian scholar, Steinman in 1973. DC regulates and maintains the immune response. There are two types of DCs, myeloid dendritic cells (MDCs) that are derived from myeloid stem cells via GM-CSF stimulation, and are termed DC1. The other type is the lymphoid dendritic cells(LDCs) or plasmacytoid dendritic cells (PDCs) that are derived from lymphoid stem cells and are termed DC2. The majority of DCs in the human body are in an immature state but have a strong ability to phagocyte antigens, while mature DCs express high levels of co-stimulators and adhesion factors. Studies have demonstrated that the co-stimulatory molecules (CD80/b7-1, CD86/b7-2, CD40) on the surface of DC in severe AA patients are higher compared to healthy individuals. The highly expressed co-stimulatory molecules provide the second signal for T cell activation, initiation of the immune response, and reduce immune tolerance [Citation34].
4.2 . Natural killer cells (NK cells)
NK cells are vital immune cells in the body. Through innate and antibody-dependent cell-mediated cytotoxicity (ADCC), NK cells play an important role in antiviral infection and immune surveillance. The proportion of NK cells in severe AA patients were found to be reduced significantly, while immunosuppressive therapy restored NK cell numbers [Citation35].
4.3. T lymphocytes and their secreted cytokines
Immune disorders induced by AA are mainly due to the cellular hyperimmune state. T lymphocytes are the main effector cells in the immune system. Abnormal T cell subsets and changes in the levels of negative regulatory factors play an important role in the occurrence and development of AA.
It has been demonstrated that the number of CD8+ cytotoxic T cells in the bone marrow and the peripheral blood of AA patients is higher [Citation36,Citation37]. Due to T cell receptor (TCR) restriction, the clonal amplification of autoimmune CD8+ cytotoxic T cells results in increased secretion of pro-inflammatory factors, including interferon-gamma(IFN-γ) and tumor necrosis factor α(TNF-α). This in turn synergistically induces apoptosis of CD34+ cells via Fas/FasL interaction [Citation38–41]. In addition to CD8+ cytotoxic T cells, CD4+T cells play an important role during AA. CD4+ T cells differentiate into Th1 cells, leading to an increase in IFN-γ levels. CD4+ T cells also differentiate into IL-4-producing CD4+ T cells (Th2 cells), IL-17-producing CD4+ T cells (Th17 cells) and regulatory T cells(Tregs). Shahram Kordasti et.al examined 63 AA patients and demonstrated that the levels of Th1 and Th2 cells in AA patients were higher compared to healthy individuals. In addition, the number of Tregs in patients with severe AA was lower compared to healthy and non-severe AA patients, while the number of Th17 cells was increased in patients with severe AA. It was demonstrated that Th1 cells were clonally restricted using spectra typing and high-throughput deep sequencing, and hence may be antigen-driven to damage Tregs. Different subgroups of Tregs have varying functions, for example, CD4+CD25+CD45RA-Foxp3low cells secrete pro-inflammatory cytokines IL-17, IL-2 and IFN-γ, which have inhibitory roles in AA [Citation41].
IFN-γ and TNF-α levels in the bone marrow of AA patients are significantly higher compared to healthy individuals [Citation42–44]. IFN-γ plays an important role in both the innate and adaptive immunity and is a negative regulator of stem and precursor cell proliferation and survival [Citation45]. They are produced by activated T cells in the bone marrow and have a profound impact on the hematopoietic system. IFN-γ can inhibit the production of several hematopoietic cell types, such as B cells [Citation46], red blood cells [Citation47], eosinophils [Citation48] and neutrophils [Citation49]. In comparison to healthy controls, the majority of AA patients had a T to A single nucleotide polymorphism at position +874 of intron 1 in the IFN-γ gene, which leads to a high expression of IFN-γ [Citation50–52]. Sharpe AH et.al found that IFN-γ could induce PD-L1 expression on T cells, NK cells, macrophages, myeloid cells and epithelial cells, which binds to PD-1 to induce apoptosis. Simultaneously, high expression of IFN-γ can induce the expression of Fas in CD34+ cells in the bone marrow. This results in the destruction of bone marrow HSCs, as well as stimulates T cells to produce TNF-α and RANKL. This in turn leads to bone marrow hematopoietic failure. Furthermore, Howard A.Young et.al demonstrated that IFN-γ functionally impaired and decreased common myeloid progenitor cells (CMP), granulocyte macrophage progenitor cells (GMP) and megakaryocyte-erythroid progenitor cells (MEP) proliferation. This in turn impacted hematopoiesis and resulted in an ‘empty’ marrow. Surprisingly, several studies have shown that AA patients could benefit from IST with IFN-γ neutralization treatment, implying that IFN-γ maybe a therapeutic target [Citation53]. TNF-α plays a pivotal role in the occurrence of inflammatory diseases such as diabetes, septic shock and rheumatoid arthritis [Citation54]. It is a negative regulator of hematopoiesis. Neal S Young et al. demonstrated that TNF-α – / – aplastic anemia mice were resistant to bone marrow destruction induced by allogeneic LN cell infusion and suggested that TNF-α was closely associated with apoptosis in AA [Citation55]. In addition, studies have demonstrated that IFN-γ induces TNF-α production in mouse macrophages through IFN regulatory factors, IFN-1 and IFN-8. This further implies the co-stimulation regulatory network between TNF-α and IFN-γ during the bone marrow destruction process [Citation56]. Negative regulators, such as IL-2, IL-6 and IL-10 were also observed to be significantly increased in SAA patients [Citation57], while hematopoietic positive regulators such as IL-3 and IL-11 were decreased [Citation58,Citation59].
5. Genetic background
Genetic factors play an important role in the pathogenesis of aplastic anemia, such as somatic cell mutations, telomerase gene mutations and genetic susceptibility.
5.1. Genetic susceptibility
A number of studies have reported that several human leukocyte antigen(HLA) alleles are associated with AA [Citation3]. HLA genes are located on chromosome 6p2.13 and encode the major histocompatibility complex proteins in human. Numerous studies have suggested that the specificity of HLA alleles makes the human body susceptible to AA. Zaineb Akram et.al. examined the HLA alleles of 74 AA patients using polymerase chain reaction(PCR) and serological techniques and found that compared to healthy individuals, DRB1*15(56.8%) and DQB1*06(70.3%) frequency was higher in AA patients. Based on multiple studies, DRB1*15, DRB1*03, DQB1*0601 and DQB1*0603 were found to be either susceptible or protective alleles. AA patients with DRB *1501 were found to have a better response to cyclosporine treatment [Citation3,Citation60]. CD8 of Cytotoxic T lymphocytes binds to the α-3 domain of HLA class Ⅰ to recognize auto-antigens that are present on HSC, which then subsequently initiates bone marrow failure [Citation61]. Hiroyuki Maruyama et.al used high-sensitivity flow cytometry to survey the presence of HLA-A allele-lacking leukocytes in 144 AA patients. They found that 18 of 71 (25.4%) newly diagnosed patients and 25 of 73 (34.2%) previously treated AA patients had HLA-A allele-lacking leukocytes. These strongly suggest that HLA is involved in the pathogenesis of AA [Citation62]. González-Galarza FF et.al determined that the frequency of HLA-B*40:02 was higher in Asian healthy controls, i.e. 7.9% in Japanese, 2.0% in Chinese, and 8.7% in South Koreans compared to only 1.6% in Germans and 1.8% in Italians. This may explain the higher incidence of AA in Asians compared to Caucasians [Citation63].
5.2. Clonal hematopoiesis and somatic mutations
AA is more complex disease than expected for a simple immune-mediated marrow failure. Complications include paroxysmal nocturnal hemoglobinuria (PNH) and MDS/AML and may not be initially diagnosed as an immune-mediated disorder [Citation64]. 60–75% AA patients had hematopoietic recovery after IST, however some AA patients would relapse due to the reemergence of the original oligoclonal T cells, and sometimes along with new clonal populations. Clonal hematopoiesis is common in AA. High-throughput sequencing has revealed the complexity of clonal hematopoiesis in AA patients. PNH results from a clonal expansion of cells derived from an HSC carrying a somatic mutation in the PIGA gene [Citation65]. 15–25% of AA patients treated with IST had PNH. Tichelli A et.al found that the incidence of MDS/AML after IST increased by 5–15% after 5–11.3 years [Citation66].
Clonal hematopoiesis often manifests as somatic mutations. About one third of AA patients had mutations in candidate genes for MDS, AML, or both as determined using targeted deep-sequencing, SNP array karyotyping, or whole-exome sequencing. Yoshizato T et al. investigated 156 patients with AA using targeted sequencing and found that 36% of these patients had multiple somatic mutations ranging from 1 to 7 mutations. The majority of mutations were BCOR and BCORL1 (in 9.3% of patients), PIGA (in 7.5%), DNMT3A (in 8.4%), and ASXL1 (in 6.2%) [Citation67]. Patients with PIGA, BCOR and BCORL1 mutations had a better response to IST, with improvements in progression free survival (PFS) and overall survival (OS). This implied a protective mechanism from immune-mediated destruction by pathogenic T cells [Citation68,Citation69]. However, patients with DNMT3A, ASXL1, JAK2/JAK3 or RUNX1 mutations had a poor response to IST and lower overall survival[Citation70,Citation71]. This suggests that monitoring clonal hematopoiesis and understanding the different types of mutations using deep sequencing and SNP array karyotyping are helpful to guide treatment strategies for AA patients.
5.3. Telomeres
A common clinical manifestation of AA is the presence of short telomeres in peripheral blood cells, especially in neutrophils [Citation1]. Both inherited and acquired AA are associated with anomalous short telomeres. Telomeres are specialized nucleoprotein structures located at the termini of vertebrate chromosomes. They consist of tandem repeat sequences (TTAGGG in vertebrates) bound by a 6-protein complex (TRF1, TRF2, TIN2, RAP1, TPP1, and POT1) known as shelterin [Citation72]. Telomeres protect chromosome integrity, but their lengths shorten with time. Telomerase can synthesize DNA at the ends of chromosomes to extend telomere lengths thus maintaining cell proliferation. However, over a period of time, telomere length is reduced. This subsequently results in reduced cell proliferation, eventually leading to apoptosis. It has been observed that telomere length in AA patients is reduced at a much faster pace. This leads to decreased expression levels of cell cycle checkpoint genes, such as CDK6, CDK2, MYB and MYC. Telomerase enzyme activity is decreased with simultaneous higher mutational frequencies observed in telomerase reverse transcriptase(TERT) [Citation73,Citation74]. Christian Bar et.al administered adeno-associated virus (AAV) 9 gene therapy vectors carrying the telomerase TERT gene to AA mouse models that had short telomere length. They found that telomerase activation reversed AA and improved survival [Citation1]. These findings indicated that telomerase activation could be a novel therapeutic strategy to treat AA associated with short telomere length. Hence determining telomerase activity and telomere length will be helpful for the clinical diagnosis and treatment of AA [Citation75].
6. Conclusion
AA is a bone marrow dysplasia disease induced by hematopoietic progenitor cell damage. Severe AA is defined as bone marrow cellularity of less than 25%, or 25–50% with less than 30% residual hematopoietic cells, and at least two of the following: (1) neutrophil count <0.5 × 109/L, (2) platelet count <20 × 109/L, or (3) reticulocyte count <20 × 109/L [Citation33]. Once AA is diagnosed, the severity of the disease should be determined and treated as soon as possible. HSCT and IST are the first-line treatment strategies for SAA patients. However the high cost of treatment, the lack of HCST donors and the recurrence rate after IST have led to an unsatisfactory treatment outcome.
Additional studies using epidemiology, basic and clinical research to carefully analyze the etiology and clinical pathology of AA are required. In addition, individualized treatment strategies to appropriately treat patients with AA are required to improve their prognosis, while simultaneously paying attention to the patients’ quality of life. Standardized clinical management and nursing programs could have a beneficial effect on patients lives.
Acknowledgements
The authors like to thank the department of Hematology, Affiliated Hospital of Nantong University, Nantong University for their assistance during the preparation of this manuscript. H.L. and L.W. provided the conception to review aplastic anemia, and L.W. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bär C, Povedano JM, Serrano R, et al. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood. 2016;127:1770–1779. doi: 10.1182/blood-2015-08-667485
- Fan R, Wang W, Wang XQ, et al. Incidence of adult acquired severe aplastic anemia was not increased in Shanghai, China. Ann Hematol. 2011;90:1239–1240. doi: 10.1007/s00277-011-1168-5
- Akram Z, Ahmed P, Kajigaya S, et al. Epidemiological, clinical and genetic characterization of aplastic anemia patients in Pakistan. Ann Hematol. 2019;98:301–312. doi: 10.1007/s00277-018-3542-z
- Li SS, Hsu YT, Chang C, et al. Incidence and treatment outcome of aplastic anemia in Taiwan-real-world data from single-institute experience and a nationwide population-based database. Ann Hematol. 2019;98:29–39. doi: 10.1007/s00277-018-3486-3
- Nakagawa MM, Rathinam CV. Constitutive activation of the canonical NF-kappaB pathway leads to bone marrow failure and induction of erythroid signature in hematopoietic stem cells. Cell Rep. 2018;25:2094–2109. e2094. doi: 10.1016/j.celrep.2018.10.071
- Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048
- Sertorio M, Du W, Amarachintha S, et al. In vivo RNAi screen unveils PPARgamma as a regulator of hematopoietic stem cell homeostasis. Stem Cell Rep. 2017;8:1242–1255. doi: 10.1016/j.stemcr.2017.03.008
- Maciejewski JP, Selleri C, Sato T, et al. A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture-initiating cells) in acquired aplastic anemia. Blood. 1996;88:1983–1991.
- Scopes J, Daly S, Atkinson R, et al. Aplastic anemia: evidence for dysfunctional bone marrow progenitor cells and the corrective effect of granulocyte colony-stimulating factor in vitro. Blood. 1996;87:3179–3185.
- de Bruin AM, Demirel O, Hooibrink B, et al. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood. 2013;121:3578–3585. doi: 10.1182/blood-2012-05-432906
- Ismail M, Gibson FM, Gordon-Smith EC, et al. Bcl-2 and Bcl-x expression in the CD34+ cells of aplastic anaemia patients: relationship with increased apoptosis and upregulation of Fas antigen. Br J Haematol. 2001;113:706–712. doi: 10.1046/j.1365-2141.2001.02810.x
- Maciejewski JP, Selleri C, Sato T, et al. Increased expression of Fas antigen on bone marrow CD34+ cells of patients with aplastic anaemia. Br J Haematol. 1995;91:245–252. doi: 10.1111/j.1365-2141.1995.tb05277.x
- Callera F, Falcao RP. Increased apoptotic cells in bone marrow biopsies from patients with aplastic anaemia. Br J Haematol. 1997;98:18–20. doi: 10.1046/j.1365-2141.1997.1532971.x
- Scopes J, Bagnara M, Gordon-Smith EC, et al. Haemopoietic progenitor cells are reduced in aplastic anaemia. Br J Haematol. 1994;86:427–430. doi: 10.1111/j.1365-2141.1994.tb04761.x
- Timeus F, Crescenzio N, Doria A, et al. Flow cytometric evaluation of circulating CD34+ cell counts and apoptotic rate in children with acquired aplastic anemia and myelodysplasia. Exp Hematol. 2005;33:597–604. doi: 10.1016/j.exphem.2005.02.005
- Holmberg LA, Seidel K, Leisenring W, et al. Aplastic anemia: analysis of stromal cell function in long-term marrow cultures. Blood. 1994;84:3685–3690.
- Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/S0092-8674(00)80258-5
- Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040
- Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612
- Yoon KA, Cho HS, Shin HI, et al. Differential regulation of CXCL5 by FGF2 in osteoblastic and endothelial niche cells supports hematopoietic stem cell migration. Stem Cells Dev. 2012;21:3391–3402. doi: 10.1089/scd.2012.0128
- Wu L, Mo W, Zhang Y, et al. Impairment of hematopoietic stem cell niches in patients with aplastic anemia. Int J Hematol. 2015;102:645–653. doi: 10.1007/s12185-015-1881-2
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905
- Majumdar MK, Thiede MA, Haynesworth SE, et al. Human marrow-derived mesenchymal stem cells (MSCs) express hematopoietic cytokines and support long-term hematopoiesis when differentiated toward stromal and osteogenic lineages. J Hematother Stem Cell Res.. 2000;9:841–848. doi: 10.1089/152581600750062264
- Gao F, Chiu SM, Motan DA, et al. Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death Dis. 2016;7:e2062. doi: 10.1038/cddis.2015.327
- Bacigalupo A, Valle M, Podesta M, et al. T-cell suppression mediated by mesenchymal stem cells is deficient in patients with severe aplastic anemia. Exp Hematol. 2005;33:819–827. doi: 10.1016/j.exphem.2005.05.006
- Chao YH, Peng CT, Harn HJ, et al. Poor potential of proliferation and differentiation in bone marrow mesenchymal stem cells derived from children with severe aplastic anemia. Ann Hematol. 2010;89:715–723. doi: 10.1007/s00277-009-0892-6
- Chao YH, Tsai C, Peng CT, et al. Cotransplantation of umbilical cord MSCs to enhance engraftment of hematopoietic stem cells in patients with severe aplastic anemia. Bone Marrow Transplant. 2011;46:1391–1392. doi: 10.1038/bmt.2010.305
- Gonzaga VF, Wenceslau CV, Lisboa GS, et al. Mesenchymal stem cell benefits observed in bone marrow failure and acquired aplastic anemia. Stem Cells Int. 2017;2017:8076529. doi: 10.1155/2017/8076529
- Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519. doi: 10.1182/blood-2006-03-010777
- Chen J, Desierto MJ, Feng X, et al. Immune-mediated bone marrow failure in C57BL/6 mice. Exp Hematol. 2015;43:256–267. doi: 10.1016/j.exphem.2014.12.006
- Young NS. Hematopoietic cell destruction by immune mechanismsn acquired aplastic anemia. Semin Hematol. 2000;37:3–14. doi: 10.1016/S0037-1963(00)90026-X
- Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European group for blood and marrow transplantation (EBMT). Haematologica. 2007;92:11–18. doi: 10.3324/haematol.10075
- Chuncharunee S, Wong R, Rojnuckarin P, et al. Efficacy of rabbit antithymocyte globulin as first-line treatment of severe aplastic anemia: an Asian multicenter retrospective study. Int J Hematol. 2016;104:454–461. doi: 10.1007/s12185-016-2053-8
- Grossmayer GE, Munoz LE, Gaipl US, et al. Removal of dying cells and systemic lupus erythematosus. Mod Rheumatol. 2005;15:383–390. doi: 10.3109/s10165-005-0430-x
- Liu C, Li Z, Sheng W, et al. Abnormalities of quantities and functions of natural killer cells in severe aplastic anemia. Immunol Invest. 2014;43:491–503. doi: 10.3109/08820139.2014.888448
- Barnes DW, Mole RH. Aplastic anaemia in sublethally irradiated mice given allogeneic lymph node cells. Br J Haematol. 1967;13:482–491. doi: 10.1111/j.1365-2141.1967.tb00758.x
- Hinterberger W, Rowlings PA, Hinterberger-Fischer M, et al. Results of transplanting bone marrow from genetically identical twins into patients with aplastic anemia. Ann Intern Med. 1997;126:116–122. doi: 10.7326/0003-4819-126-2-199701150-00004
- Li JP, Zheng CL, Han ZC. Abnormal immunity and stem/progenitor cells in acquired aplastic anemia. Crit Rev Oncol Hematol. 2010;75:79–93. doi: 10.1016/j.critrevonc.2009.12.001
- Giannakoulas NC, Karakantza M, Theodorou GL, et al. Clinical relevance of balance between type 1 and type 2 immune responses of lymphocyte subpopulations in aplastic anaemia patients. Br J Haematol. 2004;124:97–105. doi: 10.1046/j.1365-2141.2003.04729.x
- Kordasti S, Marsh J, Al-Khan S, et al. Functional characterization of CD4+ T cells in aplastic anemia. Blood. 2012;119:2033–2043. doi: 10.1182/blood-2011-08-368308
- Schuster FR, Hubner B, Fuhrer M, et al. Highly skewed T-cell receptor V-beta chain repertoire in the bone marrow is associated with response to immunosuppressive drug therapy in children with very severe aplastic anemia. Blood Cancer J. 2011;1:e8. doi: 10.1038/bcj.2011.6
- Dubey S, Shukla P, Nityanand S. Expression of interferon-gamma and tumor necrosis factor-alpha in bone marrow T cells and their levels in bone marrow plasma in patients with aplastic anemia. Ann Hematol. 2005;84:572–577. doi: 10.1007/s00277-005-1022-8
- Dufour C, Ferretti E, Bagnasco F, et al. Changes in cytokine profile pre- and post-immunosuppression in acquired aplastic anemia. Haematologica. 2009;94:1743–1747. doi: 10.3324/haematol.2009.007815
- Hinterberger W, Adolf G, Bettelheim P, et al. Lymphokine overproduction in severe aplastic anemia is not related to blood transfusions. Blood. 1989;74:2713–2717.
- Liu H, Mihara K, Song G, et al. Interferon-gamma attenuates the survival activity of G-CSF through PI3 K/Akt signaling pathway in mouse multipotent progenitor cells. Ann Hematol. 2007;86:547–555. doi: 10.1007/s00277-007-0308-4
- Arens R, Tesselaar K, Baars PA, et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity. 2001;15:801–812. doi: 10.1016/S1074-7613(01)00236-9
- Libregts SF, Gutierrez L, de Bruin AM, et al. Chronic IFN-gamma production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood. 2011;118:2578–2588. doi: 10.1182/blood-2010-10-315218
- de Bruin AM, Buitenhuis M, van der Sluijs KF, et al. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-gamma. Blood. 2010;116:2559–2569. doi: 10.1182/blood-2009-12-261339
- de Bruin AM, Libregts SF, Valkhof M, et al. IFN-gamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood. 2012;119:1543–1554. doi: 10.1182/blood-2011-07-367706
- Dufour C, Capasso M, Svahn J, et al. Homozygosis for (12) CA repeats in the first intron of the human IFN-gamma gene is significantly associated with the risk of aplastic anaemia in Caucasian population. Br J Haematol. 2004;126:682–685. doi: 10.1111/j.1365-2141.2004.05102.x
- Fermo E, Bianchi P, Barcellini W, et al. Immunoregulatory cytokine polymorphisms in Italian patients affected by paroxysmal nocturnal haemoglobinuria and aplastic anaemia. Eur J Immunogenet Off J Brit Soc Histocompatibility Immunogenet. 2004;31:267–269. doi: 10.1111/j.1365-2370.2004.00480.x
- Serio B, Selleri C, Maciejewski JP. Impact of immunogenetic polymorphisms in bone marrow failure syndromes. Mini Rev Med Chem. 2011;11:544–552. doi: 10.2174/138955711795843356
- Lin FC, Karwan M, Saleh B, et al. IFN-gamma causes aplastic anemia by altering hematopoietic stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699–3708. doi: 10.1182/blood-2014-01-549527
- Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15:24–27. doi: 10.1038/sj.cr.7290259
- Sun W, Wu Z, Lin Z, et al. Macrophage TNF-alpha licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood. 2018;132:2730–2743. doi: 10.1182/blood-2018-05-844928
- Vila-del Sol V, Punzon C, Fresno M. IFN-gamma-induced TNF-alpha expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol (Baltimore, Md.: 1950). 2008;181:4461–4470. doi: 10.4049/jimmunol.181.7.4461
- Gidvani V, Ramkissoon S, Sloand EM, et al. Cytokine gene polymorphisms in acquired bone marrow failure. Am J Hematol. 2007;82:721–724. doi: 10.1002/ajh.20881
- Eder M, Geissler G, Ganser A. IL-3 in the clinic. Stem Cells. 1997;15:327–333. doi: 10.1002/stem.150327
- Cork BA, Tuckerman EM, Li TC, et al. Expression of interleukin (IL)-11 receptor by the human endometrium in vivo and effects of IL-11, IL-6 and LIF on the production of MMP and cytokines by human endometrial cells in vitro. Mol Hum Reprod. 2002;8:841–848. doi: 10.1093/molehr/8.9.841
- Zaimoku Y, Takamatsu H, Hosomichi K, et al. Identification of an HLA class I allele closely involved in the autoantigen presentation in acquired aplastic anemia. Blood. 2017;129:2908–2916. doi: 10.1182/blood-2016-11-752378
- Gao GF, Jakobsen BK. Molecular interactions of coreceptor CD8 and MHC class I: the molecular basis for functional coordination with the T-cell receptor. Immunol Today. 2000;21:630–636. doi: 10.1016/S0167-5699(00)01750-3
- Maruyama H, Katagiri T, Kashiwase K, et al. Clinical significance and origin of leukocytes that lack HLA-A allele expression in patients with acquired aplastic anemia. Exp Hematol. 2016;44(10):931–939. e933. doi: 10.1016/j.exphem.2016.05.013
- Gonzalez-Galarza FF, Takeshita LY, Santos EJ, et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015;43(Database issue):D784–D788. doi: 10.1093/nar/gku1166
- Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128(3):337–347. doi: 10.1182/blood-2016-01-636381
- Risitano AM, Maciejewski JP, Green S, et al. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet (London. England. 2004;364:355–364.
- Tichelli A, Schrezenmeier H, Socie G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA working party of the European group for blood and marrow transplantation. Blood. 2011;117:4434–4441. doi: 10.1182/blood-2010-08-304071
- Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47. doi: 10.1056/NEJMoa1414799
- Murakami Y, Kosaka H, Maeda Y, et al. Inefficient response of T lymphocytes to glycosylphosphatidylinositol anchor-negative cells: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2002;100:4116–4122. doi: 10.1182/blood-2002-06-1669
- Gargiulo L, Papaioannou M, Sica M, et al. Glycosylphosphatidylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121:2753–2761. doi: 10.1182/blood-2012-11-469353
- Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124:2698–2704. doi: 10.1182/blood-2014-05-574889
- Marsh JC, Kulasekararaj AG. Management of the refractory aplastic anemia patient: what are the options? Blood. 2013;122:3561–3567. doi: 10.1182/blood-2013-05-498279
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005
- Sakaguchi H, Nishio N, Hama A, et al. Peripheral blood lymphocyte telomere length as a predictor of response to immunosuppressive therapy in childhood aplastic anemia. Haematologica. 2014;99:1312–1316. doi: 10.3324/haematol.2013.091165
- Brummendorf TH, Maciejewski JP, Mak J, et al. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. 2001;97:895–900. doi: 10.1182/blood.V97.4.895
- Zeng Y, Katsanis E. The complex pathophysiology of acquired aplastic anaemia. Clin Exp Immunol. 2015;180:361–370. doi: 10.1111/cei.12605