
ABSTRACT
Introduction: RUNX1 mutations in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) are associated with distinct clinicopathologic features. However, the clinical and laboratory characteristics of the myeloid malignancies may be influenced by the presence of more concomitant mutations. The aim of this study is to provide a further understanding of mutational landscape in the context of RUNX1 mutation in AML/MDS.
Methods: The present study screened for 49 mutations using next-generation sequencing (NGS). FLT3-ITD, NPM1, and CEBPA mutations were detected by PCR Sanger sequencing.
Results: One or more co-mutations were detected in all AML and 92.3% MDS patients in the context of RUNX1 mutation. The most common co-mutation was DNMT3A, followed by NRAS, IDH1, and FLT3-ITD in AML. The four more frequently co-mutated genes were U2AF1, TET2, PTPN11, and ASXL1 in MDS. We also identified a significantly difference in co-mutational spectrums between RUNX1-mutatedAML and MDS patients, as reflected in incidence of DNMT3A (35.1% vs 7.7%), FLT3-ITD (16.2% vs 0%) and U2AF1 (10.8% vs 30.7%) mutations. RUNX1-mutated AML patients with 3, or ≥4 co-mutations showed much lower CR rate than that with 2 additional mutations (p = 0.0247, 0.00919).
Conclusion: RUNX1-mutated AML and MDS are associated with a different complex co-mutation cluster. Some co-mutations have certain influence on the clinical feature and CR rate in the context of RUNX1 mutation.
1. Introduction
The runt-related transcription factor 1 (RUNX1) gene, a member of the transcription factor family, is a critical regulator of myeloid differentiation and involved in hematopoietic stem cell emergence and regulation [Citation1,Citation2]. RUNX1 was first identified as the fusion partner of RUNX1T1 (ETO) in patients with translocation involving t(8;21) acute myeloid leukemia (AML) [Citation3]. Previously reported that rare germline RUNX1 mutations are associated with the autosomal dominant familial platelet disorder (FPD) predisposing the affected individuals to AML [Citation4]. Recently, acquired RUNX1 mutations have been found in 5–10% of de novo AML, showing prognostic adverse impact on overall survival and disease progression [Citation5,Citation6]. Several studies have demonstrated RUNX1 mutations are correlated with distinct clinicopathologic features, and inferior prognosis [Citation5,Citation6]. Based on the accumulated evidence, RUNX1-mutatedAML has been included as a new provisional entity in the revised 2016 World Health Organization (WHO) classification of human myeloid neoplasms [Citation7].
Although RUNX1 mutations are very important in promoting leukemic transformation, they are sufficient to initiate disease by themselves. Secondary mutations are required before patients develop malignancy [Citation8]. With the introduction of novel next-generation sequencing (NGS) techniques, numerous mutations have been newly identified that are correlated with the RUNX1-mutated AML. For example, those involving ASXL1 that confer a worse prognosis, whereas patients with the coexisting mutations in IDH2 had a better outcome [Citation5]. RUNX1 mutations have also been identified frequently in myelodysplastic syndromes (MDS) [Citation9]. Chen CY et al. have demonstrated that RUNX1 mutations can not only be detected early at the time of diagnosis, but also acquired during disease progression and is associated with poor prognosis in patients with primary MDS, and may play a role in the development and progression of a subset of patients [Citation10]. However, the landscape of concomitant mutations and the clinical features in AML/MDS patients remain unknown. The purpose of the present study was to provide a further understanding of co-mutations in the context of RUNX1 mutation in AML/MDS.
2. Materials and methods
2.1. Case identification
A total of 406 newly diagnosed de novo AML and 279 primary MDS patients were retrospectively analyzed from two medical institutions of hematology between April 2016 and September 2018. Patients who had received prior cytotoxic therapy, or carried a prior diagnosis of any myeloid neoplasm were excluded from this study. Within this cohort, we searched for AML (n = 37) and MDS (n = 39) patients with an RUNX1 mutation using NGS and further analyzed these cases. Of 406 AML patients, 286 cases received standard chemotherapy regimen (DA/IA, daunorubicin/idarubicin 60/10 mg/m2 d1-3, cytarabine 100 mg/m2 d1-7) for induction. 74 patients were prescribed with reduced intensity DA/IA (daunorubicin/idarubicin 45/6–8 mg/m2 d1-3, cytarabine 100 mg/m2 d1-7) regimen, and 46 patients received decitabine-based induction due to older age or underlying comorbidity. The study was approved by the local Ethics Committee and conducted in accordance with the Declaration of Helsinki.
2.2. Conventional cytogenetics
Bone marrow cells were harvested at initial diagnosis. Conventional G-banded or R-banded chromosomal analysis was performed on unstimulated 24-h and 48-h BM aspirate cultures using standard techniques. When possible, at least 20 metaphases were analyzed for each case, and the results were reported according to the International System for Human Cytogenetic Nomenclature [Citation11].
2.3. Mutation analysis
In total, 49 known or putative mutational gene targets in AML/MDS were examined for mutations in all cases from the cohort using a custom-designed 49-gene panel (Ion S5™ System, Thermo Fisher, San Diego, CA, U.S.A.), including the entire coding region of IDH1, IDH2, ETNK1, ETV6, EZH2, RUNX1, DNMT3A, FBXW7, FLT3, GATA2, IL7R, ASXL1, ASXL2, JAK1, JAK2, JAK3, BIRC3, BRAF, CALR, BCOR, BCORL1, CBL, CDKN2A, CSF3R, CSMD1, KIT, KRAS, MPL, MYD88, NOTCH1, NRAS, PAX5, PDGFRA, PDGFRB, PHF6, PIGA, SETBP1, SETD2, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, PTEN, PTPN11, U2AF1, WT1, and ZRSR2, with a median depth of 2000×, FLT3-ITD, NPM1, and CEBPA mutation were detected by PCR Sanger sequencing as previously described [Citation12–14].
2.4. Multiple RT–PCR
The cDNA synthesis and multiple RT–PCR were conducted by using SureFireRTTM Kit (Promega, USA) (AIBOJIN, Cat#06-104) and Leukemia Related Fusion Gene Detection Kit (YUANQI BIO), respectively. Multiple RT–PCR amplification was performed as 8 parallel multiplex reactions on 7500 Real-time PCR System (Applied Biosystems). Leukemia Related Fusion Gene Detection Kit included 41 gene fusions (BCR-ABL, SIL-TAL1, E2A-HIF, TEL-AML1, MLL-AF4, E2A-PBX1, AML1-ETO, MLL-AF9, PML-RARα, MLL-(AF6, AF10, ELL, ENL), PLZF-RARα, STAT5b-RARα, NPM-MLF1, TEL-PDGFRB, PIP1L1-PDGFRA, AML-MDS1/EVI1, AML1-MTG16, CBFβ-MYH11, DEK-CAN, TEL-ABL, ETV6-PDGFRA, NUP98-(HoxA13, HoxC11, HoxD13, HoxA9, HoxA11, PMX1), TEL-JAK2, MLL-(AF17, AF1q, AF1p, AFX, SEPT6), (NPM, FIP1L1, PRKAR1A, NUMA1)-RARα).
2.5. Statistical analysis
Statistical analyses were performed using the Statistical Package for Social Sciences (SPSS version 17.0; SPSS Institute, Chicago, IL, U.S.A.). Patient groups with nominal values were compared by the chi-square test. However, Fisher exact test would be used if the expected values of contingency tables were smaller than 5. Regarding abnormally distributed continuous data, Student t-test was employed for the comparison of the clinical features between disease groups, categorical and continuous variables. p-values < 0.05 were considered statistically significant.
3. Results
3.1. Clinical characteristics of RUNX1-mutated AML and MDS patients
RUNX1 mutations were found in 37 of 406 (9.1%) AML and 39 of 279 (13.9%) MDS patients, respectively. For the 37 AML patients with RUNX1 mutations, the male:female ratio was1:1.47, and the median age was 45 years (range, 18–64 years). According to French–American–British classification, there were 9, 3,7, 2, 15, and 1 patients diagnosed as M0, M1, M2, M4, M5, and undetermined types with RUNX1-mutated AML, respectively. Cytogenetic analysis was successfully performed in all RUNX1-mutated AML patients, and the two most common karyotypes were a normal karyotype (NK; 64.8%) and trisomy 8 (11.1%). Leukocyte counts have ranged from 0.71 to –184.0 × 109/L (median, 14.9 × 109/L). Hemoglobin levels have ranged from 45 to 109 g/dL (median, 85.0 g/L), and platelet counts have ranged from 8.0 to 166.0 × 109/L, with a median of 47.0 × 109/L.
Among 39 MDS patients, the median age was 50 years (range, 18–74 years). 66.7% (26/39) of patients were male. According to the 2016 revised criteria of the WHO, 1 single lineage dysplasia (MDS-SLD), 6 multilineage dysplasia (MDS-MLD), 8 excess blasts-1 (MDS-EB-1), 22 MDS-EB-2, and 2 unclassifiable (MDS-U).
The median leukocyte counts, hemoglobin levels and platelet counts were 2.63 × 109/L, 80.0 g/L, and 24.5 × 109/L, respectively. Cytogenetic analysis was performed in 37 of 39 patients, including 20 (54.05%, 20/37) with normal and 17 (45.9%, 17/37) with abnormal cytogenetics. The most common abnomal karyotypes were trisomy 8. Presenting clinical and laboratory features are outlined in and .
Table 1. Pretreatment clinical characteristics of AML patients with RUNX1 mutations.
Table 2. Pretreatment clinical characteristics of MDS patients with RUNX1 mutations.
3.2. Spectrum of co-mutations and incidence in RUNX1-mutatedAML
To investigate the spectrum of co-mutations in the RUNX1-mutated AML, 50 additional genes were picked up for a complete mutational screening in 37 AML and 39 MDS patients. After excluding sequencing/mapping errors and known or possible polymorphisms, 32 in 50 genes panel were identified as mutants. Among RUNX1-mutated AML patients, all cases demonstrated co-mutations at diagnosis, which reached an average number 2.54 (range, 0–6) per case in addition to RUNX1 mutation. The most common concomitant mutation was DNMT3A (35.1%, 13/37), followed by NRAS (24.3%, 9/37), IDH1 (18.9%, 7/37), FLT3-ITD (16.2%, 6/37), FLT3-TKD (13.5%, 5/37), TET2 (10.8%, 4/37), KIT (10.8%, 4/37), and NPM1 (10.8%, 4/37). Of note, 5 patients had a combination of RUNX1, DNMT3A, and NRAS mutations. RUNX1 mutations rarely co-occurred with biallelic CEBPA mutations (n = 3). Mutated genes were grouped into several functional groups (). The mutations involved in DNA methylation (51.3%, 19/37), receptors/kinases (43.2%, 16/37), transcription factor (40.5%, 15/37) were found frequently in RUNX1-mutated AML patients. Only 10.8% (4/37) chromosomal translocations were found, in which the involved gene fusions were CBFβ-MYH11, AML-ETO, MLL-AF6, and BCR/ABL, respectively.
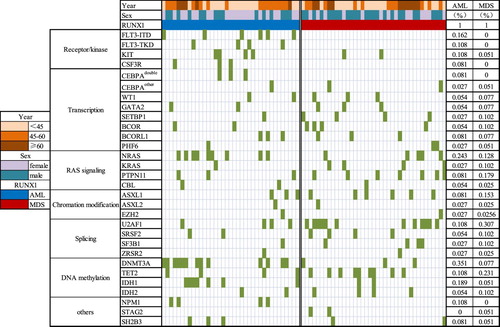
Figure 1. Landscape of co-mutations in RUNX1 mutated-AML and MDS. (a): Co-mutations in AML and MDS patients with RUNX1 mutations. Each bar represents a distinct gene. (b): Quantification of signaling pathways in RUNX1 mutated-AML and MDS. (c): Spectrum of co-mutations in AML and MDS patients divided into different mutational categories, each column represents one sample sequenced here.
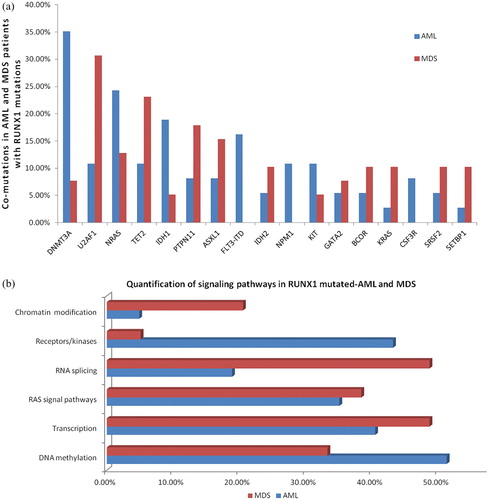
3.3. Spectrum of co-mutations and incidence in RUNX1-mutated MDS
One or more co-mutations were detected in 92.3% (36/39) RUNX1-mutated MDS. The average number of co-mutations was 2.46 (range, 0–5). The five more frequently co-mutated genes were U2AF1 (30.8%, 12/39), TET2 (23.1%, 9/39), PTPN11 (17.9%, 7/39), ASXL1 (15.4%, 6/39), and NRAS (12.8%, 5/39). Less common co-mutations (2–10.25%) involved NPM1, CEBPA, SRSF2, SF3B1, BCOR, IDH2, SRSF2, KRAS, SETBP1, DNMT3A, BCOR1, WT1, GATA2, CSMD1, IDH2, IDH1, STAG2, ETV6, and others. The most frequent functional pathway was RNA splicing and transcription, with mutations observed in as many as 48.7% of cases, followed by genes associated with RAS signal transduction pathways and DNA methylation. No gene fusions were found by multiple RT-PCR.
3.4. The difference of mutational spectrum between RUNX1-mutated AML and MDS
RUNX1-mutated AML patients had a significantly higher incidence of DNMT3A and FLT3-ITD mutations (13 of 37, 35.1%) than those with MDS (35.1% vs 7.7%, p = 0.003; 16.2% vs 0, p = 0.028), while U2AF1 mutations were observed more frequently in MDS group by comparing with AML patients (30.7% vs 10.8%, p = 0.033). All NPM1, FLT3-ITD and CSF3R mutations were found only in AML patients. There was no difference in the incidence of NRAS, TET2, IDH1/2, and KIT between patients with AML and MDS. The same was also true for KRAS, PTPN11, GATA2, BCOR, and SRSF2 mutations. AML patients mainly abnormally activate receptors/kinases signaling pathways (43.2% vs 5.1%, p = 0.000), while MDS patients focused on the RNA splicing (48.7% vs 18.9%, p = 0.006). The difference of mutational spectrum between RUNX1-mutated AML and MDS was shown in . The frequencies of detected gene mutations and gene functional groups were shown in (a/b). The spectrum of co-mutated genes is depicted in (c).
Table 3. The difference of Mutational spectrum between RUNX1-mutated AML and MDS
3.5. Analysis of the relationship between clinical characteristics and co-mutations in RUNX1-mutated AML
For the 37 AML patients with a RUNX1 mutation, the concomitant mutations in DNMT3A and NRAS were significantly associated with older age compared with wild-type AML (p = 0.000, 0.048, respectively). In contrast, the FLT3-ITD mutation was associated with significantly younger age compared with those with the FLT3 wild-type gene (p = 0.035). However, there was no significant difference (p = 0.221) between IDH1 co-mutated and IDH1 wild-type groups. The median WBC count, hemoglobin level, and platelet count were not observed in above patients.
We assessed the value of co-mutations for predicting the complete response (CR) rate after the first conventional induction chemotherapy. Of 37 AML patients enrolled, 36 cases were available for evaluating CR rate. The overall CR rate was 66.7% (24/36). The CR rate was significantly inferior in patients with a DNMT3A mutation compared with those with the DNMT3A wild-type (38.4% vs 82.6%, p = 0.00696). Patients with IDH1 mutations also show a lower CR rate compared with those without IDH1 mutations (28.5% vs 75.86%, p = 0.01721). No differences were observed between patients with NRAS mutation and wild-type (55.5% vs 70.37%, p = 0.4141), FLT3-ITD and FLT3 wild-type (50% vs 70%, p = 0.34278).
Of note, the number of co-mutations also showed a strong association with CR rate. RUNX1-mutated AML patients with one, two, three, and ≥4 additional mutations presented 83.3% (5/6), 91.7% (11/12), 50% (6/12), and 33.3% (2/6) CR rate, respectively. Patients harboring 3, or ≥4 additional mutations showed much lower CR rate than that with 2 additional mutations (p = 0.0247, 0.00919). The clinical characteristics and CR rate are shown in .
Table 4. Pretreatment clinical characteristics and CR rate of the most common co-mutations in RUNX1-mutated AML patients.
3.6. Analysis of the relationship between karyotype and additional mutations in RUNX1-mutated MDS
As illustrated in , 3 co-mutations were found more frequently in NK group (p = 0.010), while 2 co-mutations were observed more frequently in abnormal karyotype group (p = 0.069). NK patients exhibited slightly more frequency in PTPN11 mutations (30% vs 5.8%, p = 0.097), and mainly abnormally activate transcription in comparison to abnormal karyotype-patients (70.0% vs 29.4%, p = 0.0016). The distribution of U2AF1, TET2, ASXL1 mutations in NK-patients, and abnormal karyotype-patients was balanced.
Table 5. Association of additional mutations with karyotypes in RUNX1-mutated MDS.
4. Discussion
Consistent with previous reports, we found RUNX1 mutations in 9.1% of de novo AML patients and 13.9% of MDS patients [Citation6,Citation9]. RUNX1 mutation has been shown to be a significant marker for resistance to standard induction therapy and for inferior survival [Citation5,Citation6,Citation15]. However, the clinical and laboratory characteristics as well as the outcome of the different WHO leukemia entities may be influenced by the presence of more concomitant mutations. Here, we describe an analysis of the co-mutational landscape in a cohort of RUNX1-mutated AML and MDS using NGS technique to gain some new insights.
In our cohort, 100% RUNX1 mutations in AML and 92.3% MDS were accompanied by additional mutations, and there was no significantly statistical difference between the two groups in the average number of co-mutations. However, significantly different spectrum of co-mutations was demonstrated between AML-patients and MDS-patients. First,RUNX1-mutated AML cases commonly showed additional mutations in genes such as DNMT3A, NRAS, and IDH1, while MDS patients exhibited more co-mutations in U2AF1, TET2, and PTPN11. DNMT3A mutations predominated in AML patients, whereas U2AF1 mutations showed a higher incidence in MDS patients. Second, functionally, more receptors/kinases and fewer RNA splicing were involved in AML-patients in comparison to MDS patients. Third, NPM1, FLT3-ITD, and CSF3R mutations are not found in MDS patient haboring RUNX1 mutations. Gaidzik VI et al. showed that RUNX1 mutations significantly associated with mutations in ASXL1 (p < 0.0001), KMT2A (p < 0.0001), and IDH2 (p = 0.02) in AML patients [Citation5]. In the 14 samples in the Bejar et al. study that had mutations in addition to RUNX1, they were most often in TET2 (12), ASXL1 (12), EZH2 (8), and NRAS (6), and there was no overlap with mutations in TP53, JAK2, ETV6, IDH1/2, NPM1, GNAS, BRAF, PTEN, or CDKN2A [Citation16]. These data suggest that RUNX1 mutated-AML and MDS share different co-mutated genes, which may be correlated with distinct clinicopathologic features, provide prognostic information, and dictate whether leukemogenesis occurs or not [Citation17,Citation18,Citation19]. Further studies are needed to clarify how to integrate this increased knowledge of co-mutations in our understanding of AML and MDS pathogenesis and into clinical practice.
Both DNMT3A and IDH1/2 are involved in the epigenetic regulation of transcription, particularly, alterations in DNA methylation. DNMT3A mutations were found in AML ranges from 18% to 36% [Citation20,Citation21]. Many recent studies have suggested that the DNMT3A mutation correlated with older age, higher relapse rates and poorer overall survival [Citation21,Citation22]. The frequencies of IDH1 and IDH2 mutations in AML are approximately 6–16% and 8–19%, respectively [Citation23,Citation24]. A recent analysis demonstrated that IDH1 and IDH2 mutations confer an adverse effect in patients with AML lacking the NPM1 mutation [Citation25]. However, some different results from other study were noted. Gaidzik V I et al performed multivariable analyses including the interaction terms RUNX1 mut/DNMT3Amut and RUNX1mut/IDH1mut, and found a significant interaction for CR, relapse-free (RFS), event-free (EFS), and overall survival (OS), neither in younger patients nor in older patients [Citation5]. However, whether the pattern of association between DNMT3Amut and RUNX1mut, IDH1mut and RUNX1mut suggests an interplay in the prognosis of AML remains unknown. In the present study, our data showed a high co-mutation rate in DNMT3A and IDH1 in RUNX1 mutated-AML. Patients in both of RUNX1mut/DNMT3Amut and RUNX1mut/IDH1mut groups had a relatively lower CR rate than those with RUNX1mut/DNMT3Awt and RUNX1mut/IDH1wt, respectively. Additionally, CR decreased with the increasing of number of co-mutations, which suggested that the mutation number may be a new evaluation factor for achievement of CR. These findings indicated that NGS-based multi-gene sequencing can provide detailed molecular information for establishing an accurate risk stratification and clinical prognosis determination.
More recently, several studies have further suggested associations of certain mutations in MDS with specific cytogenetic abnormalities [Citation26,Citation27,Citation28]. Data from Zhang, T. et al. showed an association between der (1;7) (q10;p10) and RUNX1 mutations [Citation26]. In another NGS study of 179 MDS patients displayed that the most notable associations were between monosomal karyotype and TP53 mutations, RUNX1 and +21, and SF3B1 with inv (3)(q21q26.2) and del (11q), respectively. Other associations included ASXL1 with +8, −7/del (7q) with IDH1and U2AF1 mutations [Citation28]. Our study showed that +8 was the most frequent cytogenetic abnormality in the context of RUNX1 mutation, neither +21 nor der(1;7)(q10;p10) was found in MDS patients. This result was different from the report of Tefferi A and Zhang, T. [Citation26,Citation27]. The reason for this difference might be due to the age and gender distribution of the sample, the small sample size, or the research background of our cohort.
In conclusion, RUNX1-mutated AML and MDS are associated with a different complex co-mutation cluster, and there was a significantly different co-mutation profile between these AML and MDS patients. Some co-mutations have certain influence on the clinical feature and CR rate in AML. The major limitations of our study are derived from the absence of PFS and OS because of the different consolidation regimens administrated. Further work is necessary to achieve a better understanding of the genetic and biological basis for RUNX1 mutated-AML and MDS, and this in turn will lead to improved therapies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
ORCID
Feng Zhou http://orcid.org/0000-0002-1544-6692
Additional information
Funding
References
- Swiers G, De Bruijn M, Speck NA. Hematopoietic stem cell emergence in the conceptus and the role of Runx1. Int J Dev Biol. 2010;54(6–7):1151–1163. doi: 10.1387/ijdb.103106gs
- Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017;129(15):2070–2082. doi: 10.1182/blood-2016-10-687830
- Miyoshi H, Shimizu K, Kozu T, et al. T(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci U S A. 1991;88(23):10431–10434. doi: 10.1073/pnas.88.23.10431
- Michaud J. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood. 2002;99(4):1364–1372. doi: 10.1182/blood.V99.4.1364
- Gaidzik V I, Teleanu V, Papaemmanuil E, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016 Nov;30(11):2160–2168. doi: 10.1038/leu.2016.126
- Jalili M, Yaghmaie M, Ahmadvand M, et al. Prognostic value of RUNX1 mutations in AML: a meta-analysis. Asian Pac J Cancer Prev. 2018;Feb 26;19(2):325–329.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544
- Ripperger T, Steinemann D, Gohring G, et al. A novel pedigree with heterozygous germline RUNX1 mutation causing familial MDS-related AML: can these families serve as a multistep model for leukemic transformation? Leukemia. 2009 Jul;23(7):1364–1366. doi: 10.1038/leu.2009.87
- Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014 Feb;28(2):241–247. doi: 10.1038/leu.2013.336
- Chen CY, Lin L, Tang JL, et al. RUNX1 gene mutation in primary myelodysplastic syndrome–the mutation can be detected early at diagnosis oracquired during disease progression and is associated with poor outcome. Br J Haematol. 2007 Nov;139(3):405–414. doi: 10.1111/j.1365-2141.2007.06811.x
- Willatt L, Morgan SM, Shaffer LG, et al. ISCN 2009 an international system for human cytogenetic nomenclature. Hum Genet. 2009;126(4):603–604. doi: 10.1007/s00439-009-0726-6
- Rau R, Brown P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol Oncol. 2009 Dec;27(4):171–181. doi: 10.1002/hon.904
- Lin LI, Chen CY, Lin DT, et al. Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin Cancer Res. 2005 Feb 15;11(4):1372–1379. doi: 10.1158/1078-0432.CCR-04-1816
- Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999 May 1;93(9):3074–3080.
- Hata T. Current diagnosis and treatment for myelodysplastc syndromes. Rinsho Ketsueki. 2017;58(4):373–380.
- Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343
- Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017 Apr 13;129(15):2070–2082. doi: 10.1182/blood-2016-10-687830
- Schnittger S, Dicker F, Kern W, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011 Feb 24;117(8):2348–2357. doi: 10.1182/blood-2009-11-255976
- Mangan JK1, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit RevOncog. 2011;16(1-2):77–91.
- Sehgal AR, Gimotty PA, Zhao J, et al. DNMT3A mutational status affects the results of dose-escalated induction therapy in acute myelogenous leukemia. Clin Cancer Res. 2015 Apr 1;21(7):1614–1620. doi: 10.1158/1078-0432.CCR-14-0327
- Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. doi: 10.1056/NEJMoa1005143
- Tan M, Ng IKS, Chen Z, et al. Clinical implications of DNMT3A mutations in a Southeast Asian cohort of acute myeloi leukaemia patients. J Clin Pathol. 2017 Aug;70(8):669–676. doi: 10.1136/jclinpath-2016-204195
- Rakheja D, Konoplev S, Medeiros LJ, et al. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43(10):1541–1551. doi: 10.1016/j.humpath.2012.05.003
- DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732–736. doi: 10.1002/ajh.24072
- Yamaguchi S, Iwanaga E, Tokunaga K, et al. IDH1 and IDH2 mutations confer an adverse effect in patients with acute myeloid leukemia lacking the NPM1 mutation. Eur J Haematol. 2014;92(6):471–477. doi: 10.1111/ejh.12271
- Zhang T, Xu Y, Pan J, et al. High frequency of RUNX1 mutation in myelodysplastic syndrome patients with whole-arm translocation of der(1;7)(q10; p10). Leukemia. 2017 Oct;31(10):2257–2260. doi: 10.1038/leu.2017.228
- Tefferi A, Idossa D, Lasho TL, et al. Mutations and karyotype in myelodysplastic syndromes: TP53 clusters with monosomal karyotype, RUNX1 with trisomy 21, and SF3B1 with inv(3)(q21q26.2) and del(11q). Blood Cancer J. 2017 Dec 18;7(12):658. doi: 10.1038/s41408-017-0017-8
- Fernandez-Mercado M, Burns A, Pellagatti A, et al. Targeted re-sequencing analysis of 25 genes commonly mutated in myeloid disorders in del(5q) myelodysplastic syndromes. Haematologica. 2013 Dec;98(12):1856–1864. doi: 10.3324/haematol.2013.086686