
ABSTRACT
Objectives: A sensitive screening for the coexistence of α0-thalassemia and the hemoglobin E (Hb E) trait is important to identify at-risk couples for hydrops fetalis. However, previous cutoff values have shown a positive predictive value (PPV) of only 50% or less. This study aimed to define more specific indicators to reduce the need for DNA tests.
Methods: Patients with Hb E trait, as diagnosed by high performance liquid chromatography (HPLC) and/or isoelectric focusing (IEF) techniques, were tested for α0-thalassemia and α+-thalassemia deletions using multiplex gap polymerase chain reaction. Iron deficiency anemia (IDA) were excluded using a red cell distribution width (RDW) of more than 14.5%.
Results: From 390 specimens, suitable cutoff values showing a 100% sensitivity for detection of heterozygous α0-thalassemia were an Hb E level of less than 22% by HPLC, a mean corpuscular volume (MCV) of less than 72 fL, and a mean corpuscular hemoglobin (MCH) level of less than 22.5 pg. Comparable results were obtained in the validation cohort (N = 179). Using a combination of Hb E with either MCV or MCH cutoff points gave a PPV of 76.2% and 77.4%, respectively.
Discussion: IDA was reported to interfere with Hb E level. In this study, we excluded IDA using RDW of more than 14.5% to enhance the test specificity.
Conclusion: Lower cutoff screening values can be used to exclude α0-thalassemia in the Hb E trait yielding a higher specificity in a normal RDW condition. This can save the cost and labor of DNA testing.
Introduction
Thalassemia and hemoglobinopathy are the most common human monogenetic diseases [Citation1]. These inherited disorders of hemoglobin synthesis are characterized by a reduced production of the globin chains of hemoglobin [Citation2]. Alpha- and beta-thalassemia are the two most common subtypes of thalassemia syndromes which affect α- and β-globin chain production, respectively [Citation3]. There are two major types of α-thalassemia; α+-thalassemia and α0-thalassemia (–/αα), of which the latter is the more severe form and can lead to the homozygous fatal disease, Hb Bart's hydrops fetalis, in the offspring [Citation4,Citation5].
Approximately 7% of the world’s population are carriers of thalassemia and 300,000–500,000 babies are born with severe homozygous or compound heterozygous forms of these conditions each year [Citation6]. The two most common α0-thalassemias are –SEA and –Mediterranean, which are mostly found in Southeast Asia and the Mediterranean basin, respectively [Citation6].
In areas where both hemoglobin E (Hb E) and α0-thalassemia are prevalent, the combination of Hb E with α0-thalassemia is common. Due to the diminished availability of α-globin chains and less efficient αβ-heterodimer assembly by βE [Citation7], the double heterozygote (Hb E/α0-thalassemia) results in a marked reduction of Hb E levels.
In a previous study [Citation4], the suitable cutoff values of the Hb E level, mean corpuscular volume (MCV), and mean corpuscular hemoglobin (MCH) as screening indicators for Hb E/α0-thalassemia double heterozygotes at 100% sensitivity were reported to be a Hb E level of less than 26% (by HPLC), MCV of less than 74 fL, and MCH of less than 24 pg, giving 52.2%, 48.1%, and 49.4% specificity, respectively. Combining the Hb E level (as determined by HPLC) with either MCV or MCH cutoff points improved the specificity but the positive protective value (PPV) was only 50% and 46.7%, respectively. Moreover, no cutoff value for Hb E determined by IEF has been reported so far.
Iron deficiency anemia (IDA) causes low MCV, MCH, and Hb E levels and, therefore, may result in a false positive screening for α0-thalassemia. Previous studies have not excluded patients with IDA, even though they might affect the cutoff screening values for the detection of α0-thalassemia in the Hb E trait. However, as a high red cell distribution width (RDW) is a sensitive marker for IDA [Citation8], it could be used to exclude this confounder. In this study, we aimed to determine suitable cutoff values with higher specificity to reduce the need for DNA tests, as they are expensive and not generally available.
Materials and methods
We collected blood specimens from Hb E trait patients diagnosed by HPLC and/or IEF, between March 2017 and April 2018. Hemoglobin analysis was performed using isoelectric focusing (IEF) method with IsoScan Imaging System for Hemoglobin Testing (Perkin–Elmer Life and Analytical Sciences, Turku, Finland) and/or high-performance liquid chromatography (HPLC) (Bio-Rad Laboratories, CA) to determine the quantity of each hemoglobin type. Hb E trait was diagnosed by Hb E level greater than 10%, Hb A is present with greater than Hb E level without elevation of Hb F. Complete blood count was measured by an automated cell counter (Haematologie, Montpellier, France).
Blood samples from patients with IDA were excluded using the criteria of exclusion of a RDW of more than 14.5% (higher than a normal range) [Citation9]. We collected baseline red cell parameters, which were the MCV, RDW, and MCH. The α0-thalassemia (–SEA, –Thai, –Philippine, –Mediterranean, and –20.5Kb deletion) and α+-thalassemia determinants (3.7 and 4.2 Kb deletion) were detected by multiplex gap PCR. The cutoff parameters (Hb E level by HPLC/IEF, MCV, and MCH) to uncover co-existing α0-thalassemia were determined in a training set (N = 390) and then validated with the data from another set of patients (N = 179). This research was approved by the institutional review board of the institution.
Statistical analysis
Sensitivity, specificity, PPV, and negative predictive values (NPV) of the selected cutoff points were calculated. Red cell parameters (hemoglobin, hematocrit, MCV, and MCH) are presented as the mean ± one standard deviation (SD). Analyses of the cutoff Hb E level, MCV, and MCH were determined from the receiver operator characteristic (ROC) curve using Stata version 13.0 software (Stata Corp LP, College Station, TX) and the R program version 3.3.2.
Results
Blood specimens were collected from a total of 450 patients with the Hb E trait from March 2017 to April 2018 at the Thalassemia laboratory in the King Chulalongkorn Memorial Hospital (Bangkok, Thailand). A total of 60 samples were excluded from the study due to having either a RDW of more than 14.5% (N = 40), and so likely to be IDA, or the patients age being less than 18 years old (N = 20). Baseline parameters of the remaining 390 blood specimens included in this study are shown in . All specimens were tested for hemoglobin typing by IEF and 285 (73.1%) specimens were also tested by HPLC.
Table 1. Baseline parameters of the specimens.
Among the 390 Hb E trait specimens, heterozygous α0-thalassemia was detected in 49 (12.6%) specimens (12.3% of –SEA and 0.25% of –Thai deletion), while heterozygous α+-thalassemia was detected in 88 (22.6%) samples, comprised of 53 heterozygous 3.7 kb deletion (13.6%), five heterozygous 4.2 kb deletion (1.3%), and 30 homozygous 3.7 kb or 4.2 deletions (7.7%).
The data were then analyzed to determine a suitable cutoff value to detect co-inherited heterozygous α0-thalassemia in the Hb E trait using a likelihood ratio at a 95% confidential interval (CI) and ROC curve. To detect heterozygous α0-thalassemia, an Hb E level of less than 22% by HPLC was found to achieve 100% sensitivity, 88.5% specificity, 64.9% PPV, and 100% NPV, while a Hb E level of less than 37.3% by IEF achieved 100% sensitivity with 67% specificity, 32% PPV, and 100% NPV. A MCV of less than 72 fL achieved 100% sensitivity, 70.2% specificity, 32% PPV, and 100% NPV, while a MCH of less than 22.5 pg achieved 100% sensitivity, 85.7% specificity, 49.5% PPV, and 100% NPV in detecting the co-inheritance heterozygous α0-thalassemia with Hb E trait (). The ROC curves are shown in .
Figure 1. The ROC curve of Hb E 22% (HPLC), 37.3% (IEF), MCV 72 fL, and MCH 22.5 pg for detection of heterozygous α0-thalassemia in the Hb E trait.
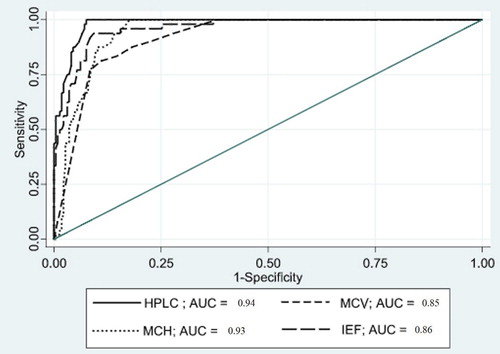
Table 2. Sensitivity, specificity, PPV, and NPV for detecting a heterozygous α0-thalassemia in patients with a HbE trait for each cutoff value.
Improvements in the specificity and PPV for the detection of co-inherited α0-thalassemia with Hb E trait were found when combining two of these parameters (). Using a Hb E level of less than 22% (by HPLC) with either a MCV of less than 72 fL or a MCH of less than 22.5 pg revealed 100% sensitivity, 95.6% specificity, 100% NPV, and 76.2% or 95.6% PPV, respectively.
Table 3. Sensitivity, specificity, PPV, and NPV for detecting heterozygous α0-thalassemia in patients with a Hb E trait using a combination of two parameters.
For the validation cohort, we utilized the other 179 blood specimens (Hb E trait and a RDW of less than 14.5%) to confirm the diagnostic accuracy, and compared the results with the training set using a paired t-test at a cutoff Hb E level of 22% (HPLC)/37.3% (IEF), MCV of 72 fL, and MCH of 22.5 pg. There was no significant difference between the training and validation sets (Supplementary Table 1). The baseline parameters of the validation set are shown in .
Discussion
In this study, all samples with heterozygous Hb E with α0-thalassemia demonstrated Hb E levels lower than 22% (HPLC) or 37.3% (IEF), a MCV of less than 72 fL, and a MCH of less than 22.5 pg. These cutoff points showed 100% sensitivity for screening heterozygous α0-thalassemia in the Hb E trait. The values were lower than in a previous study that reported suitable cutoff values of a Hb E of less than 25% (HPLC), MCV of less than 74 fL, and MCH of less than 24 pg [Citation4]. When applying these previous cutoff levels to re-analyze our training data (n = 390), the obtained PPV values were much lower at 30.1%, 21.1%, and 23%, respectively (Supplementary Table 2). Thus, using the new cutoff values of this study showed a marked improvement in the PPV to 64.9%, 32%, and 49.5%, respectively. Supporting our data, another study demonstrated that a Hb E level of less than 21.54% was the best cut-point value for screening the coexistence of heterozygous α0-thalassemia (–SEA/) with the Hb E trait [Citation10].
It was reported that IDA decreases the Hb E levels and correction of IDA restored the amount of Hb E [Citation11]. The mean RDW value in the previous study [Citation4] was 17.6%, which might have resulted in these included cases causing false–positive results in the screening. In this study, we excluded patients with a RDW of more than 14.5%, in order to exclude IDA and so enhance the test specificity. Using our cutoff values in cases with a normal RDW will help to reduce unnecessary PCR tests to confirm the coexistence of heterozygous α0-thalassemia. For patients with concomitant IDA, iron supplement may restore the Hb E levels and obviating the need for DNA tests.
Although determination of the Hb E level in adult patients by IEF for screening of the coexistence of heterozygous α0-thalassemia with the Hb E trait has never been reported, in this study we found a Hb E of 37.3% or more, as determined by IEF, showed 100% sensitivity and 100% NPV.
The strength of this study was that the independent training and validation data sets were consistent with each other. However, the limitation was that there was no available data on the serum ferritin level for definitive diagnosis of IDA. Nevertheless, ferritin assays are not routinely performed in these patients as, in practice, serum ferritin analysis is only performed in cases with a high RDW.
In conclusion, our data showed that the cutoff values of a Hb E level of less than 22% by HPLC, or less than 37.3% by IEF, a MCV of less than 72 fL, and a MCH of less than 22.5 pg should be used to detect the co-inherited heterozygous α0-thalassemia in Hb E trait with a normal RDW. The specificity and PPV can be further improved by using the combination of the Hb E level (by HPLC) with either the MCV or MCH.
Supplemental Material
Download (27.5 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Weatherall DJ, Clegg JB. Thalassemia - a global public health problem. Nat Med. 1996;2:847–849. doi: 10.1038/nm0896-847
- Weatherall DJ. The thalassaemias. Br Med J. 1997;314:1675–1678. doi: 10.1136/bmj.314.7095.1675
- Olivieri NF. The beta-thalassemias. N Engl J Med. 1999;341:99–109. doi: 10.1056/NEJM199907083410207
- Sanchaisuriya K, Chirakul S, Srivorakun H, et al. Effective screening for double heterozygosity of Hb E/ α0-thalassemia. Ann Hematol. 2008;87(11):911–914. doi: 10.1007/s00277-008-0520-x
- Weatherall DJ, Clegg JB. The α thalassaemias and their interactions with structural haemoglobin variants. In: Weatherall DJ, Clegg JB, editors. The Thalassaemia syndromes. 4th edn. Oxford: Blackwell Science; 2001. p. 484–525.
- Christianson A, Howson CP, Modell B. The hemoglobin disorders: sickle cell anemia and thalassemia. In: Christianson A, Howson CP, Modell B, editors. March of Dimes – Global report on birth defect. New York: White Plains; 2006. p. 24–25.
- Wong SC, Ali MA. Hemoglobin E diseases: hematological, analytical, and biosynthetic studies in homozygotes and double heterozygotes for alpha-thalassemia. Am J Hematol. 1982;13:15–21. doi: 10.1002/ajh.2830130104
- Guyatt GH, Patterson C, Ali M, et al. Diagnosis of iron-deficiency anemia in the elderly. Am J Med. 1990;88:205–209. doi: 10.1016/0002-9343(90)90143-2
- Evans TC, Jehle D. The red blood cell distribution width. J Emerg Med. 1991;9:71–74. doi: 10.1016/0736-4679(91)90592-4
- Pornprasert S, Treesuwan K, Punyamung M, et al. Hba2/E level found in Co-inheritance with the α thalassemia -1–SEA/ type deletion and either Hb E or β-thalassemia. Hemoglobin. 2012;36(4):381–387. doi: 10.3109/03630269.2012.679375
- Fucharoen S, Winichagoon P. Clinical and hematologic aspects of hemoglobin E ß-thalassemia. Curr Opin Hematol. 2000;7:106–112. doi: 10.1097/00062752-200003000-00006