
ABSTRACT
Objective: The outcomes of alternative donor hematopoietic stem cell transplantation (HSCT) with unmanipulated grafts for Inherited bone marrow failure syndromes (IBMFS) are discouraging. Our study is to demonstrate that IBMFS with disease-specific characteristics requires a tailored conditioning regimens to enhance engraftment and reduce regimen related toxicities. Methods: We retrospectively analyzed 42 patients diagnosed with IBMFS and transplanted with an alternative donor graft at our center from November 2012 to August 2018. Twenty-seven patients had Fanconi anemia (FA), 7 had dyskeratosis congenita (DC), and 8 had severe congenital neutropenia (SCN). Patients received ex-vivo unmanipulated alternative donor grafts from a matched unrelated donor (MUD) (n = 22), haploidentical donor (HID) (n = 17) and unrelated cord blood donor (UCBD) (n = 3). FA and DC patient subgroups received reduce intensified conditioning (RIC), while SCN patients received a myeloablative conditioning (MAC) regimen. Results: The median follow-up time for the surviving patients was 38 months (range: 9–63 months). The failure-free survival (FFS) for entire cohort was 76.1%, and was 72.4%, 100% and 56.2% for patients with FA, DC and SCN, respectively. There were no primary graft failures. The cumulative incidence of aGVHD at day 100 was 48.1%. The cumulative incidence of cGVHD at 1 and 3 years was 35.0% and 69.3%, respectively. Conclusion: HSCT using alternative donors with unmanipulated grafts and disease-specific conditioning regimens for IBMFS patients shows promising survival.
Introduction
Hematopoietic stem cell transplantation (HSCT) still represents the only curative option for patients with inherited bone marrow failure syndromes (IBMFS), although transplant does not correct the congenital malformations or lower the risk of the emergence of solid tumors due to treatment [Citation1–3]. Most IBMFS patients do not have HLA-identical unaffected (i.e. Fanconi anemia negative) sibling donors and, therefore, require alternative (HLA-mismatched related or unrelated) donor sources. In recently years, improved outcomes have been reports with grafts from alternative donors for patients with FA, but most HSCTs were performed using ex vivo T-cell depleted (TCD) grafts. The number of studies using alternative donors, particularly haploidentical donors (HID) with unmanipulated grafts remains limited and results are less encouraging [Citation4]. Graft rejection, a high incidence of graft versus host disease (GVHD) and regimen-related toxicity are the main causes of treatment failure [Citation5]. Defective DNA repair mechanisms in Fanconi anemia (FA), telomere length dysregulation in dyskeratosis congenita (DC), and maturation arrest of myeloid precursors due to one of a number of genetic mutations can all result in bone marrow failure and as a result recurrent infectious complications prior to HSCT [Citation6,Citation7]. Transplant-related mortality (TRM), especially opportunistic infections, likely increase among IBMFS patient undergoing an allo-HSCT [Citation8]. Therefore, disease-specific HSCT using an optimized conditioning regimen based on the characteristics of each disease is likely essential for achieving long-term survival for IBMFS patients [Citation8].
In this retrospective study, we present the clinical outcomes of 42 patients with IBMFS who received HSCT with disease-specific conditioning regimens using alternative, unmanipulated donor grafts.
Methods
Patients and donor selection
We respectively analyzed data on 42 patients diagnosed with IBMFS and who underwent alternative donor HSCT at the Lu Daopei Hospital between November 2012 and December 2018. All patients were confirmed to have bone marrow failure (BMF). In addition to a detailed physical examination, imaging studies, a typical physical feature assessment and family history evaluation, we also perform confirmatory tests including chromosome breakage testing using mitomycin C (MMC) and deipoxybutane (DEB), and single gene or panel-based genetic testing. All transplant eligible patients underwent bone marrow biopsy with MDS/leukemia-related cytogenetics and mutational analysis to identify evidence of clonal abnormalities, myelodysplasia or leukemic transformation. Patients also had repeated disease-specific deleterious-related gene mutation analyses [Citation6].
Indications for HSCT for patients with FA and DC were progressive BMF with pending or overt transfusion dependence, development of myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML)/cytogenetic abnormalities. For patients with SCN, indication for HSCT were uncontrolled recurrent life-threatening infections and median absolute neutrophil count [ANC] consistently less than 2.19 × 109/L despite >8ug/kg daily r-HuG-CSF treatment [Citation9]. The study protocol was approved by the ethics committee at the Hebei Yanda Lu Daopei Hospital. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
All related donors had genetic counseling and confirmatory genetic testing that they did not carry any deleterious congenital genetic mutations. For the patients who did not have a suitable matched identical sibling donor available, HID was used if a matched unrelated donor (MUD) was unavailable. Donor-specific HLA antibodies (DSA) were measured routinely pretransplant in all patients, the results were analyzed as mean fluorescence intensity (MFI) versus donor specific mismatch. If MFL ≥ 10,000, we need to changed the donor source due to associated with an extremely high risk of graft rejection. When there was no suitable MUD or HID available, an unrelated cord blood donor (UCBD) was used.
Definitions and Evaluation
Neutrophil engraftment was defined as the first day that the absolute neutrophil count was ≥0.5 × 109/L for three consecutive days and platelet recovery was defined as the first day that the platelet count was ≥20 × 109/L for seven days without platelet transfusion. No patient received granulocyte colony stimulating factor (G-CSF) post-HCT to accelerate neutrophil recovery. Both acute graft versus host disease (aGVHD) and chronic graft versus host disease (cGvHD) were defined and graded according to previously published criteria [Citation10,Citation11]. Transplant-related mortality (TRM) was defined as any death not due to recurrence of disease. Failure free survival (FFS) was defined as survival with sustained engraftment, whereas death, graft-rejection and late graft-rejection were categorized as treatment failures. MDS and AML were defined according to the WHO 2012 definition.
Disease specific conditioning regimen and related organ toxicity
The reduced Intensity conditioning (RIC) regimen for FA patients was busulfan (Bu; total dose of 6.4 mg/kg, IV) d-7–6, Fludarabine (FLU; total dose of 120 mg/m2, IV) d-5–4 Cyclophosphamide (CTX; total dose of 2.0 g/m2, IV) d-5–4. For DC patients, TBI d-6 (total 300cGy, was delivered side-to-side, instead of anterior–posterior, with the patient in supine position and the arms resting at the side of the thoracic cage to allow partial pulmonary compensation to reduce lung injury), FLU (total dose of 120 mg/m2, IV) d-5–2,CTX (total dose of 3.0 g/m2, IV) d-5–2. For SCN patients, myeloablative conditioning (MAC) consisted of Ara-C 9–10 g/ m2, Bu (total dose of 12.8 mg/kg, IV,) d-9–6, CTX (total dose of 3.6 g/m2 IV) d-5−4. All patients received a combination either of two different ATG:rabbit ATG (ATG-T) [Sanofi,Paris, France], rabbit anti-human thymocyte immunoglobulin, total dose 7.5–10 mg/kg divided four days in 26 cases or ATG-F [Astellas Pharma Inc, Tokyo, Japan], rabbit anti-human lymphocyte immunoglobulin, total dose 20 mg/kg divided four days in 14 cases. Campath-1 [Sanofi,Paris, France], anti-CD52 mAb (total dose 1mg/kg divided five days in 2 cases). Organ toxicity was graded according to the Common Terminology Criteria for Adverse Events version 5.0 (2018) for stem cell transplantation recipients (https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_5.0/).
Stem cell source
HLA typing of the patient and donor was performed at the allele level for HLA-A, B, C and DRB1,DQB1 in the case of PBSCs. In the case of umbilical cord blood, HLA typing was performed at the Ag level for HLA-A and B and at the allele level for DRB1. Unmanipulated stem cells were used for all patients. The 17 patients in the HID-HSCT cohort received granulocyte colony-stimulating factor (G-CSF)-primed BM combined with peripheral blood stem cells (PBSCs). The 22 patients in the MUD HSCT cohort received G-CSF-mobilized PBSCs (the Chinese Marrow Donor Program only provide G-CSF-mobilized PBSCs in China). The total target mononuclear cells were ≥5 × 108/kg and CD34+ cells ≥2 × 106/kg of the recipient weight in HID and MUD cohorts. Three patients in the UCBD cohort received one unit CB. Each cord unit had more than 4 × 107 nucleated cells/kg.
GVHD prophylaxis, management, and supportive care
All patients received tacrolimus (target concentration of 5–10 ng/mL), mycophenolate mofetil (0.5 g i.v every 12h in adults and 0.25 g i.v. every 12 h pediatric patients, from conditioning regimen to 30 days post-HSCT), and short-term methotrexate (15 mg/m2 on day +1, 10 mg/m2 on days +3, +6, and +11) as GVHD prophylaxis. The tapering of calcineurin inhibitors started at nine months post-transplant in the absence of obvious GVHD and continuous full donor chimerism (FDC). For the treatment of aGVHD, the effective concentration of tacrolimus was resumed, and 1–2 mg/kg/day methylprednisolone equivalent was administered as a first-line therapy. For steroid-refractory aGVHD, second-line treatments such as an anti-CD25 monoclonal antibody (Basiliximab, Novartis, Switzerland) or tumor necrosis factor-α antibody (rhTNFR:Fc, CPGJ, China) were administered. The viral surveillance screening for cytomegalovirus (CMV) using PCR was conducted two times per week. CMV DNA ≥ 500 copies/mL in plasma was defined as positive CMV activation. Ganciclovir (dehydroxphenylglycol) or foscarnet therapy was initiated for CMV activation.
Analysis of chimerism
Chimerism was assessed by short tandem repeat analysis in fractionated BM CD33+ cells and peripheral blood CD3+ T-cells[Citation12]. Chimerism analysis was performed at days 28, 60, and 90 and then every 6 months. Analysis was also performed when there were significant changes in blood counts or development of new cytopenia. FDC was defined as the presence of at least 95% donor hematopoietic cells, whereas mixed donor chimerism was defined as 5% to 95% donor cells post-transplantation.
Statistical methods
The last follow-up for all surviving patients was on July 30, 2019. The probabilities of FFS was analyzed using the Kaplan-Meier estimator and the survival differences between subgroups were tested by log-rank test for statistical significance. The cumulative incidences of GVHD were estimated in the competing risk model with death as the competing event. For univariate analysis, age, sex, transfusion requirement (20 units), family history, chromosome breakage testing positivity, congenital abnormalities, presence of deleterious complementation group mutations, pre-transplant treatment, donor types and cell doses were analyzed, Univariate and multivariate analyses were done to determine whether any of the selected factors were predictive of the clinical endpoints. All factors with P < .10 in the univariate analysis were evaluated in the Cox regression model with a backward stepwise model selection approach. Significant factors (P < .05, two-sided tests) were considered as independent predictive factors of outcomes. The statistical analysis was performed using SPSS version 19.0 (SPSS Inc, Chicago, IL).
Results
Baseline characteristics
The general characteristics of the FA patients (n = 27) and donors are summarized in . The median age at diagnosis and HSCT were 5 years (range: 1m–25y) and 11 years (range: 3y–26y), respectively. Male to female ratio was 17:10. Thirteen (48.1%) patients had congenital malformations. Seven of them presented absent or abnormal thumbs and Cafe au lait spots, five had short stature and developmental delay, four had congenital heart disease, or kidney or genital abnormalities. Twenty-six percent of cases (n = 7) had a family history of congenital malformations Eight patients tested positive for chromosomal fragility and all of the other patients tested negative. Disease-deleterious genotypes were present in all FA patients: FANCA mutations were present in 55.5% (n = 15), FANCC in 14.8% (n = 4), FANCG in 7.4% (n = 2), BRCA2 in 3.7% (n = 1) and NHP2 in 3.7% (n = 1). Twenty-two of FA patients (88.8%) were initially misdiagnosed as acquired aplastic anemia (AA) and were treated with androgens or cyclosporine prior to HSCT. All patients received prior red blood cell or platelet transfusions and 20 (74.0%) patients had transfusion dependence pre-HSCT. Twenty-three patients had serve cytopenia and three of these patients developed MDS while two patients developed AML. The median time from diagnosis to transplant was 2.3 years (range: 1m–9y). Sixteen patients received MUD-HSCT and 7 patients received HID-HSCT.
Table 1. Baseline characteristics of patients with fanconi anemia undergoing transplantation.
The general characteristics of the DC patients and donors are summarized in and . The male to female ratio was 6:1, the median age at diagnosis and HSCT were 4 years (range: 4y–22y) and 10 years (range: 5y–26y). Two patients had a family history of congenital malformations, two had typical characteristics of the triad of reticular skin pigmentation, nail dystrophy, and oral leukoplakia and one had congenital heart disease. Progressive BMF and deleterious telomerase-associated gene mutations were present in all patients. Three patients had TINF2 mutations, 1 patient each had a mutation in either TERT, WRAP56, CTC1 or RTEL1. Five patients received cyclosporine and androgen treatment before HSCT, all patients were transfusion-dependent at the time of HSCT, and no patient developed MDS or AML prior to HSCT.
Table 2. Baseline characteristics of patients with dyskeratosis congenital.
Table3. Donor and transplantation characteristics for dyskeratosis congenital patients.
The general characteristics of the SCN patients and donors are summarized in and . The male to female ratio was 5–3. The median age at diagnosis and HSCT was 2 months (range: 1 week–1 year) and 3.1 years (range: 2.1 years–15 years). The median disease course pre-HSCT was 3 years (range: 8 months–15 years). The median absolute neutrophil count (ANC) in PB was 150 (20–300)/L. Two patients had a family history – one with a congenital megacolon and one with development retardation. All patients had uncontrolled recurrent infection prior to HSCT. Lobectomy was performed in one patient due to severe pulmonary infection. One patient had colonic resection due to repeated infection of a congenital megacolon. ELANE deleterious genes mutations were detected in all patients. Two patients had mutations that were traced back to their parents. All patients received G-CSF treatment for a median of 36 months (range: 24 m–120 m) before HSCT and doses above the median dose of 8 μg/kg/day (range: 5–16 μg/kg/day).
Table 4. Baseline Characteristics of Patients with Severe Congenital Neutropenia.
Table 5. Donor and transplantation characteristics for patients with severe congenital neutropenia.
Conditioning related to organ toxicity and engraftment and chimerism analysis
No Grade 4 regimen-related toxicity was observed, but five patients (2 FA, 2 DC, 1 SCN) experienced Grade 3 mucositis and six patients (3 FA, 2 DC, 1 SCN) experienced transient grade 2 liver transaminases. No primary graft failure was observed. The median neutrophil engraftment time was 14 days (range: 10–21 days), there were no difference among the three different disease types (P = 0.451) or three different donor types (n = 0.531). Forty patients achieved platelet engraftment. The median engraftment time was 13 days (range: 10–45 days). There were no differences among the three different disease types (P = 0.631), but there was a prolonged engraftment time for UCB of 25 days (range: 22–45 days) compared to 13 days (range: 8–23 days) for HID and 14 days (range: 9–27 days) for MUD (P = 0.012). Chimerism results were available for all patients. The median time for performing the first donor chimerism was 20 days (range: 15–28 days) post-transplantation. Thirty-five patients had 100% donor chimerism, four patients had donor chimerism between 95% and 100%. Three patients initially had a mixed chimerism (90–95%), but subsequently converted to full donor chimerism. By 3 months, all patients achieved full donor chimerism except one patient who developed secondary graft failure at 15 months and died.
Graft versus host disease
Grade 2–4 aGVHD at day 100 occurred in 13 cases of FA, 2 cases of DC and 2 cases of SCN. There were no differences based on the donor type: 36.3% for MUD, 47.0% for HID and 33.3% for UCB (P = 0.516). There were also no differences by disease type: 50.0% FA,25.0% DA, 25.0% SCN (P = 0.272). In multivariate analysis, a positive chromosomal fragility test was the only risk factor (P = 0.000) related to aGVHD by day 100 in the FA group. Neither aGVHD (P = 0.130) nor cGVHD (P = 0.666) had a significance impact on FFS in the FA group. Chronic GVHD occurred in 10 FA patients (extensive in 3 patients), 3 DC patients (extensive in 1 patient) and 3 SCN patients (extensive in 1 patient). The cumulative incidence of 3-year cGVHD was higher in the UCB cohort 78.5% vs 40.4% in the MUD and 57.1% in the HID cohort (P = 0.003). There were no differences among the FA (53.3%), DC (47.7%) and SCN (50.0%) disease groups (P = 0.795). Among 27 patients who survived for more than 1 years, only four patients needed low dose immunosuppression treatment (IST) to control cGVHD, while all other patients were able to successfully stop all IST. In multivariate analysis UCB was the only risk factor correlated with development of cGVHD [P = 0.008, 2.286 (HR 1.237–4.233)].
Infection complications
The cumulative incidence of day 100 cytomegalovirus (CMV) viremia was 64.7%, the median time to CMV reactivation was 33 days (range: 26–57 days) post-HSCT. There were no differences based on donor type: MUD (54.4%), HID (76.4%), UCB (66.6%) (P = 0.326). There were also no differences by disease group: FA (63.6%), DA (57.1%), SCN (75.0%) (P = 0.416). The cumulative incidence of day 100 Epstein–Barr virus (EBV) viremia was 38.0%. The median time for EBV activation was 38 days (range: 16–78 days) post-HSCT. There were no differences by donor type: MUD (36.3%), HID (47.0%) and UCB (33.3%) (P = 0.772). There were also no differences by disease group: FA (40.7%), DA (42.8%) and SCN (25.0%) (P = 0.812). One patient developed post-transplantation lymphoid tissue proliferative disease (PTLD) that responded to IST cessation, anti-CD20 antibody treatment, and donor lymphocyte infusion (DLI) but died of intracranial bleeding. Three patients with FA had sepsis during the first first month post-transplantation. Six patients developed a new lung infection. Four of seven DC patients developed new lung complications after HSCT (one with small airway lesion in the right lower lobe; one with pulmonary GVHD; two with mixed bacterial and fungal infection). All patients with SCN had recurrent infection prior to HSCT. One patient had a lobectomy due to severe pulmonary infection and developed a lung infection again after HSCT. One patient underwent colonic resection due to repeated infection of a congenital megacolon before HSCT and developed recurrent infection at the site of a fistula after HSCT. Five patients developed a new infection (4 lung and 1 intestinal) post-HSCT.
Survival outcomes and follow-up
The median follow-up time for survivors was 38 months (range: 9–63 months), the FFS for the entire cohort was 74.8%, and was 73.3%, 100% and 56.2% for patients with FA, DC and SCN, respectively (P = 0.173) (). There were no differences in FFS among the MUD (71.1%), HID (76.4%) and UCB (100%) cohorts (P = 0.715). In the univariate and multivariate analyses, a positive chromosome fragility test was the only Independent adverse prognostic factor on survival outcomes for the FA patients [P = 0.022, 0.224 (HR 062–.802)] (). A total of 10 patients died. The main causes of death were GVHD in 5 patients (50%; 4 aGVHD, 1 cGVHD), central neutral system infection in 1 patient (20%), intracranial bleeding in 1 patient (10%), drug-induced encephalopathy in 1 patient (10%), secondary graft failure in 1 patient (10%) and mucor infection in the ENT site in 1 patientt (10%). There were no secondary malignancies observed after HSCT at the last follow up.
Figure 1. Kaplan–Meier estimate of event-free survival for patients with Fanconi anemia, dyskeratosis congenita and severe congenital neutropenia
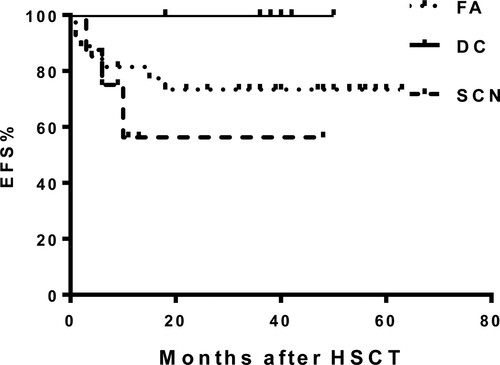
Table 6. Univariate analysis of parameter in FA group.
Discussion
In our study, alternative donor and disease-specific conditioning regimens in an unmanipulated HSCT setting for IBMFS resulted in positive effects on survival. Among all patients, the 3 year FFS was 76.1% and 72.4%, 56.2%, and 100% in FA, DC, and SCN patients, respectively. There are limited studies on the use of alternative donors, especially unmanipulated haploidentical grafts for FA patients [Citation13]. Allo-HSCT for dyskeratosis congenita is characterized by a marked decline in long-term survival due to delayed deaths from pulmonary complications, with alternative donor sources appearing to be poor prognostic markers associated with early mortality [Citation14]. HLA-mismatched donor allo-HSCT for SCN showed inferior 3-year OS of 55% compared to 85% among HLA-matched and unrelated HLA-matched donors [Citation15]. The frequency of graft failure was about 10% in the largest ever reported cohort of 136 FA patients received alternative donor-HSCT [Citation15]. In our IBMFS cohort, most of the patients were transfusion-dependent and proper diagnosis was delayed as some were initially diagnosed with acquired aplastic anemia and treated with prolonged immunosuppression therapy prior to referral for HSCT. Our study cohort did not have matched sibling donors and all infused alternative stem cell donor sources were unmanipulated. We opted to use disease-specific conditioning regimens to better cross the HLA barrier, promote engraftment and minimize life-threatening disease-specific complications, which we reasoned would translate into better FFS.
In the ex-vivo CD34-selected/T-cell–depleted graft setting, alternative donor-HSCT for FA patients have significantly decreased early TRM and GVHD, showed promising OS in the range of 70–89%, but have a higher incidence of graft failure of about 12% [Citation13]. There are limited studies on using alternative donors especially HID for FA in the unmanipulated graft setting [Citation16]. Early experience with HID-HSCT for FA revealed dismaying outcomes of 53.8% 5-year OS [Citation17]. Fludarabine-based and radiation-free preparative regimens improved engraftment and long-term OS based on many previously published studies [Citation4Citation18], and should be the standard regimen for patients with FA undergoing alternative donor HSCT. However, the low dose CTX/FLU alone may not be a sufficient conditioning therapy as there is a substantial number of residual host cells initially following transplant and a higher risk of engraftment failure [Citation19]. EBMT reported the probability of 8% of primary graft failure with a FLU-based conditioning regimen for HLA-matched unrelated donor HSCT. In our current FA subgroup, all patients were alternative donors (MUD in 17 cases and HID in 9 cases). Most of the patients were heavily pretreated with transfusions, CSA and androgen or ATG, both of which have been identified as independent risk factors for engraftment failure and TRM after HSCT [Citation20,Citation21]. We reasoned that addition of low-dose busulfan, an alkylating agent with low immunosuppressive activity, could have a synergistic effect with cyclophosphamide to enhance engraftment [Citation22]. Hence, we developed a low-dose Bu (0.8 mg/kg, IV,d-7,-6), FLU (30 mg/m2, IV, d-5,-2) and low dose CTX (500mg/m2, IV, d-5,-2) based-RIC conditioning regimen with the goal of promoting stem cell engraftment. Only two of four patients with progression to MDS or AML received Bu for four days (total 3.2 mg/kg). There was no primary graft failure with this chemotherapy based conditioning regimen. All achieved full donor chimerism at three months post-HSCT. Only one patient developed secondary graft failure at 15 months post-HSCT and died. There were no differences in median neutrophil engraftment between the MUD and HID cohorts. Thus, our regimen with the addition of low dose of busulfan to flu-ATG, was safe, well-tolerated with excellent engraftment and survival outcomes.
In univariate and multivariate analyses, a positive chromosomal fragility test was the only independent adverse prognostic factor that negatively affected FFS, which is in line with a previous report [Citation23]. We did not find that age [Citation20,Citation24], donor type [Citation18], presence of congenital misformation [Citation25] or progression to MDS/AML [Citation26] were negative predictive factors in our study as has been previously reported. Although a chromosomal fragility test provides a reliable marker for the diagnosis of FA, not all patients exhibit sensitivities to DNA-crosslinking agents such as DEB or MMC, so diagnosis of FA now relies on detection of deleterious FA genes and progressive BMF [Citation27,Citation28]. Dysfunctional DNA breakage repairment associated with FA results in marked hypersensitiveness to alkylating agents and radiation [Citation29]. In our cohort, the patients with a positive chromosomal fragility test had a higher incidence of TRM and adversely affected FFS. Interestingly, the positive chromosomal fragility test also correlated with increased incidence of aGVHD, but, in our FA cohort, this increase did not have significant impact on the overall FFS.
Allo-HSCT is the only curative treatment for patients with DC. However, transplant cannot correct other systemic defects due to impairment of telomere length maintenance. A transplant procedure can accelerate telomere shortening thereby increasing early transplant-related toxicities [Citation30]. Hypersensitivity to irradiation and chemotherapy, and possible pre-existing organ dysfunction result in a poor survival rate following a conventional myeloablative conditional regimen (MAC) [Citation31]. Fludarabine containing non-MAC regimens are being increasingly used and will hopefully lead to better outcomes [Citation32]. There are no standard HSCT protocols available for DC patients with clinical outcomes worse when alternative donors are used compared with a sibling donor HSCT [Citation33]. Given that previous studies showed FLU (total 200 mg/m2)/CTX (50 mg/kg–1500 mg/m2), ATG, TBI200cGy RIC facilitated successful hematopoietic cell engraftment in adults with dyskeratosis congenital [Citation34,Citation35], we developed a low-dose FLU (total dose 100–120 mg/m2) and CTX (total dose 2000 mg/m2) – ATG(7.5–10 mg/kg) based RIC. Considering that our DC patients had two risk factors for increased graft failure: an alternative donor (4 MUD and 3 HID) and transfusion dependence, we also added TBI 300cGy to our conditioning regimen. Since pulmonary complications are one of the major causes of late death after HSCT, the low dose TBI in our regimen was delivered side-to-side, instead of anterior- posterior, with the patient in a supine position and the arms resting at the side of the thoracic cage to allow partial pulmonary compensation to reduce lung injury. no patients developed pulmonary fibrosis or secondary malignancies until the last follow-up time.
The conditioning regimen for patients with SCN are usually a busulfan and cyclophosphamide-based MAC regimen [Citation36,Citation37]. This combination is effective and allows achievement of full chimerism in almost all patients. However, the standard conditioning regimen and GVHD prophylaxis have not been established. EBMT analyzed the largest SCN transplant cohort reported of 136 patients with an 82% 3-year OS and 17% TRM [Citation15]. Most (87%) of the patients received a MAC regimen. The engraftment rate was similar to that from RIC, but no chimerism was reported [Citation15] Oshima et al reported that reduced conditioning regimens and a history of massive blood transfusions were risk factors for graft rejection even in a matched sibling donor transplantation setting [Citation36]. Infection complications, aGVHD and increased age have also been associated with poor outcomes [Citation37]. We developed a MAC consisting of Ara-C (9 g/m2), Bu (12.8 mg/Kg), Flu (150–200 mg/m2) or CTX (3.6 g/m2) (HID only), ATG (10 mg/kg), MeCCNU (250 mg/m2) (MUD only). In our cohorts, all patients were engrafted, although patient who received UCB had a delayed neutrophil engraftment. Six of eight patients achieved full chimerism and two initially had a mixed chimerism but achieved full donor chimerism by three months post-HSCT. At the last follow-up, three patients died. The causes of death were gut aGVHD (MUD) at 3 months post-HSCT, drug-induced encephalopathy (HID) at 6 months post-HSCT and CNS bleeding (MUD) at 10 months post-HSCT.
All patients had recurrent infection before HSCT. One patient received a lobectomy and another had a colonic resection due to resistant severe uncontrollable infection. Five patients developed a new site of infection post-HSCT. With a median follow-up of 14 months (range: 6–48 months), five patients remained alive without secondary malignancies and long-term complications. Our FFS is inferior to the one reported by the EBMT. Except for those patients that received cord blood from donors, all of our SCN patients received peripheral blood grafts and most donors were HID. Both cord blood and peripheral stem cell sources have been link to inferior EFS outcomes [Citation38].
FA patients have defective DNA repair mechanisms and therefore their tissue repair and recovery is relatively slowly following treatment with a conditioning regimen which may increase their risk of developing aGVHD. GVHD is an important factor impacting the outcome of an allo-HSCT for IBMF, especially because GVHD is an inflammatory state that can lead to microsatellite instability and genomic alterations within epithelial tissues, predisposing a patient to the development of cancer [Citation4]. In our cohort, Grade 2–4 aGVHD at day 100 occurred in 13 patients with FA, 2 patients with DC and 2 patients with SCN. cGVHD occurred in 10 patients with FA (extensive in 3 patients), 3 patients with DC (extensive in 1 patient) and 3 patients with SCN (extensive in 1 patient). This is higher than the 10% to 20% rate of aGVHD reported and 10% cGVHD rate reported when ex-vivo CD34-selected/T-cell–depleted graft are used [Citation18]. But our rates of aGVHD and cGVHD are comparable to those reported in transplants using in vivo- T-cell-depleted grafts [Citation8]. Patients who received UBC grafts had a higher cumulative 3-year cGVHD incidence, 78.5%, compared to a cumulative incidence of 40.4% for those with a MUD and 57.1% for those with a HID (P = 0.003). A UBC graft source was the only risk factor related to higher cumulative incidence of cGVHD in our study. It was not surprising that the main cause of death was GVHD in 5 patients (50%; 4 aGVHD,1 cGVHD) in our cohorts. We still need to optimize our GVHD prophylaxis regimen to minimized GVHD incidence in our transplant patients. Infection was another important factor that impacted outcome in IBMFS patients and was the second most common cause of death in our study.
Modification of the conditioning regimen based on the type of IBMFS not only can address disease-specific safety and toxicity issues, but can also lead to better long-term clinical outcomes. Encouragingly, In our alternative donor, unmanipulated graft setting, we did not observe any Grade 4 regimen-related toxicity, secondary malignancies or post-transplant comorbidities. Patient with DC did especially well with our novel conditioning regimen combination. There are some limitations in our study. This is a retrospective study, the number of patients in our study is relatively small and the length of follow-up is limited. Strategies such as better prophylaxis for infection control, inclusion of cyclosporine as part of GVHD prophylaxis to lower rates of GVHD, more frequent post-HSCT pulmonary function assessments for early pulmonary dysfunction detection will be important to implement. Future studies that aim to reduce infection complications and incidence of GVHD are warranted to further improve the outcomes for IBMFS patients.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Macmillan ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia – when and how? Br J Haematol. 2010;149(1):14–21.
- Alter BP. Inherited bone marrow failure syndromes: considerations pre- and posttransplant. Hematology Am Soc Hematol Educ Program. 2017;1:88–95.
- Sieff CA. Introduction to acquired and inherited bone marrow failure. Hematol Oncol Clin North Am. 2018;32(4):569–558.
- Mehta PA, Davies SM, Leemhuis T, et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood. 2017;129(16):2308–2315.
- Dalle J-H, de Latour RP. Allogeneic hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Int J Hematol. 2016;103(4):373–379.
- Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol. 2017;177(4):526–542.
- Alter BP, Giri N, Savage SA, et al. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018;103(1):30–39.
- Li Q, Luo C, Luo C, et al. Disease-specific hematopoietic stem cell transplantation in children with inherited bone marrow failure syndromes. Ann Hematol. 2017;96(8):1389–1397.
- Peffault de Latour R, Peters C, Gibson B, et al. Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Bone Marrow Transplant. 2015;50(9):1168–1172.
- Harris AC, Young R, Devine S, et al. International, multicenter standardization of acute graft-versus-host disease clinical data collection: a report from the Mount Sinai acute GVHD international consortium. Biol Blood Marrow Transplant. 2016;22(1):4–10.
- Jagasia MH, Greinix HT, Arora M, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease. I. The 2014 diagnosis and staging working group report. Biol Blood Marrow Transplant. 2015;21(3):389–401.e1.
- Clark JR, Scott SD, Jack AL, et al. Monitoring of chimerism following allogeneic haematopoietic stem cell transplantation (HSCT): technical recommendations for the use of short tandem repeat (STR) based techniques, on behalf of the United Kingdom National External Quality assessment service. Br J Haematol. 2015;168(1):26–37.
- Zecca M, Strocchio L, Pagliara D, et al. HLA-Haploidentical T cell-depleted allogeneic hematopoietic stem cell transplantation in children with Fanconi anemia. Biol Blood Marrow Transplant. 2014;20:571–576.
- Kojima S, Ehlert K. Reconsidering the indication of haematopoietic stem cell transplantation for dyskeratosis congenita. Br J Haematol. 2018;183(1):11–12.
- Fioredda F, Iacobelli S, Biezen A, et al. Stem cell transplantation in severe congenital neutropenia: an analysis from the European society for blood and marrow transplantation. Blood. 2015;126(16):1885–1892.
- Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in Fanconi anemia: current evidence, challenges and recommendations. Expert Rev Hematol. 2017;10(1):81–97.
- Simon E, Zuccolo S, Dufort G, et al. Feasibility and outcome of haploidentical SCT in pediatric high-risk hematologic malignancies and Fanconi anemia in Uruguay. Bone Marrow Transplant. 2012;47(5):663–668.
- Smetsers SE, Smiers FJ, Bresters D, et al. Four decades of stem cell transplantation for Fanconi anaemia in the Netherlands. Br J Haematol. 2016;174(6):952–961.
- Maury S, Prete A, Locasciulli A, et al. Fludarabine, cyclophosphamide and anti-thymocyte globulin for alternative donor transplants in acquired severe aplastic anemia: a report from the EBMT-SAA Working Party. Bone Marrow Transplant. 2005;36(11):947–950.
- Macmillan ML, Defor TE, Young JH, et al. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood. 2015;125(24):3798–3804.
- Ebens CL, Defor TE, Tryon R, et al. Comparable outcomes after HLA-matched sibling and alternative donor hematopoietic cell transplantation for children with Fanconi anemia and severe aplastic anemia. Biol Blood Marrow Transplant. 2018;24:765–771.
- Dulley FL, Vigorito AC, Aranha FJP, et al. Addition of low-dose busulfan to cyclophosphamide in aplastic anemia patients prior to allogeneic bone marrow transplantation to reduce rejection. Bone Marrow Transplant. 2004;33(1):9–13.
- Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi anemia registry: Relation of clinical symptoms to diepoxybutane sensitivity. Blood. 1989;73(2):391–396.
- De Latour P, Porcher R, Dalle J, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European group for blood and marrow Transplantation experience. Blood. 2013;122(26):4279–4286.
- Guardiola P, Pasquini R, Dokal I, et al. Outcome of 69 allogeneic stem cell transplantations for Fanconi anemia using HLA-matched unrelated donors: a study on behalf of the European group for blood and marrow transplantation. Blood. 2010;95(2):422–429.
- Peffault de Latour R, Soulier J. How I treat MDS and AML in Fanconi anemia. Blood. 2016;127(24):2971–2979.
- Dufour C. How I manage patients with Fanconi anaemia. Br J Haematol. 2017;178(1):32–47.
- West AH, Churpek JE. Old and new tools in the clinical diagnosis of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2017;1:79–87.
- Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in Fanconi anemia: current evidence, challenges and recommendations. Expert Rev Hematol. 2017;10(1):81–97.
- Wynn RF, Cross MA, Hatton C, et al. Accelerated telomere shortening in young recipients of allogeneic bone-marrow transplants. Lancet. 1998;351(9097):178–181.
- Amarasinghe K, Dalley C, Dokal I, et al. Late death after unrelated-BMT for dyskeratosis congenita following conditioning with alemtuzumab, fludarabine and melphalan. Bone Marrow Transplant. 2007;40(9):913–914.
- Fioredda F, Korthof ET, Van Biezen A, et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br J Haematol. 2018;183(1):110–118.
- Barbaro P, Vedi A. Survival after hematopoietic stem cell transplant in patients with dyskeratosis congenita: systematic review of the literature. Biol Blood Marrow Transplant. 2016;22(7):1152–1158.
- Dietz AC, Orchard PJ, Baker KS, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46(1):98–104.
- Otrock ZK, Bacigalupo A, Mahfouz RA, et al. A reduced intensity conditioning regimen of fludarabine, cyclophosphamide, antithymocyte globulin, plus 2 Gy TBI facilitates successful hematopoietic cell engraftment in an adult with dyskeratosis congenita. Bone Marrow Transplant. 2012;47(9):1254–1255.
- Oshima K, Hanada R, Kobayashi R, et al. Hematopoietic stem cell transplantation in patients with severe congenital neutropenia: an analysis of 18 Japanese cases. Pediatr Transplant. 2010;14(5):657–663.
- Ferry C, Ouachée M, Leblanc T, et al. Hematopoietic stem cell transplantation in severe congenital neutropenia: experience of the French SCN register. Bone Marrow Transplant. 2005;35(1):45–50.
- Fioredda F, Iacobelli S, van Biezen A, et al. Stem cell transplantation in severe congenital neutropenia: an analysis from the European society for blood and marrow transplantation. Blood. 2015;126(16):1885–1892.