
ABSTRACT
Ethnic differences in haematologic malignancies remain poorly elucidated, hence research in this area is important.
This was a retrospective study into potential ethnic disparity in the presentation and outcomes of acute promyelocytic leukaemia (APL) between New Zealand (NZ) Polynesian and European patients. Data were analysed for patients treated at Auckland City Hospital (ACH; n = 55) and recorded in the New Zealand Cancer Registry (NZCR; n = 173), both for the period 2000–2017.
We found that Polynesian patients treated at ACH presented at a younger age than European (P = 0.005), showed higher blast counts (P = 0.033), and a marginally higher prothrombin ratio (P = 0.02). Treatment with all-trans retinoic acid (ATRA) was started faster in Polynesian patients than European (P = 0.021), suggesting Polynesians were sicker at presentation but were managed accordingly. There were no differences in bleeding events, transfusion requirements and early deaths during the first month of treatment. Long-term survival was also similar. Data extracted from the NZCR confirmed NZ Polynesian patients with APL were younger than European (P < 0.001), but long-term survival was similar (P = 0.920).
In summary, this study indicates a discrepancy in the presentation and severity of APL between NZ Polynesian and European patients but treatment initiation was rapid with no difference in outcomes. The distinctive features of APL in NZ Polynesians raise the possibility of a predisposing genetic factor or a different risk factor profile, elucidation of which is important for all patients with APL.
1. Introduction
Acute promyelocytic leukaemia (APL) is distinguished by the t(15;17)(q24;q21) chromosomal translocation involving retinoic acid receptor alpha (RARA) gene (on chromosome 17) and promyelocytic leukaemia (PML) gene (on chromosome 15) [Citation1]. The resulting PML-RARA fusion protein recruits co-repressors to silence regulators of myeloid differentiation [Citation2]. Morphologically, leukaemic promyelocytes are typically hypergranular but a microgranular variant accounts for 15%–20% of APL cases and can be harder to diagnose [Citation3]. APL patients often present with laboratory evidence of disseminated intravascular coagulation (DIC) and hyperfibrinolysis, which leads to haemorrhagic complications and in severe cases, death [Citation4]. Despite a high risk of early complications, current treatment regimens incorporating all-trans retinoic acid (ATRA) and arsenic trioxide, achieve complete remission and long-term survival in >90% patients [Citation5].
Rates of APL vary between ethnicities. In Los Angeles (USA), Latinos had a higher frequency of APL when compared to non-Latinos, 24.3% and 8.3% respectively of acute myeloid leukaemia (AML) cases [Citation6]. A similar difference was shown amongst Latinos outside USA; APL accounts for 20%–35% of AML in Latin American nations compared to 5%–13% in Western nations [Citation7–9]. While the specific cause for these differences remains unknown, it was suggested it could be due to genetic predisposition [Citation6]. In support, Latino patients have a significantly higher frequency of the BCR1 breakpoint than non-Latinos (78% and 52% respectively) [Citation10]. High frequency of APL is also seen in Chinese populations, reported at 19% of AML in adult patients [Citation11], and increasing to 26%–34% in paediatric patients [Citation11–13]. In contrast, in children from USA, Central and Northern Europe, APL accounts for 5%–7% of AML [Citation14]. Overall, in Western nations, APL arises most frequently in younger adults between the ages of 20–59 years, and is less often in children and elderly populations [Citation7].
In New Zealand, Māori and Pacific Island (Polynesian) ethnic populations make up 22.3% of the total population of approximately 5 million [Citation15]. Significant health inequities exist between New Zealand (NZ) Polynesians and Europeans [Citation16]. Polynesians have higher incidence and mortality from a number of cancers, including lung, breast, and stomach, compared with European patients [Citation17–19]. As for acute leukaemia, literature that explores ethnic disparities is limited. Nevertheless, compared to NZ Europeans, the age-adjusted relative risk (RR) of AML was higher in Māori children (aged 0–14 years; RR 1.84) [Citation20], Māori young adults (aged 25–49 years; RR 1.5), and Māori older adults (aged 50–74 years; RR 1.31) [Citation21]. Publication from the NZ Ministry of Health demonstrates that during 1991–2004 NZ Polynesians had a higher risk of dying from leukaemia overall (International Classification of Diseases-10 [ICD-10] codes C91–C93), with the excess mortality rate ratio 1.25 relative to Europeans [Citation16]. During a similar time period (1987–1997), NZ Polynesian patients eligible for bone marrow transplantation were less likely to find a potential 6/6 matched unrelated haematopoietic stem cell donor than European patients (P < 0.042) [Citation22]. There is no specific information on the features and outcomes of APL amongst New Zealand ethnic groups.
We recently reported the clinical presentation and outcomes of APL patients treated at Auckland City Hospital (ACH) during 2000–2017 [Citation23,Citation24]. The current study examines these data in the context of ethnicity. Our primary aim was to evaluate how presenting disease characteristics, time to treatment, and outcomes compared between NZ Polynesian and European patients. We further aimed to verify ethnic differences identified in the Auckland cohort using national data extracted from the New Zealand Cancer Registry (NZCR).
2. Patients and methods
2.1. Study cohort and data collection
Medical records for APL patients diagnosed and managed at ACH were retrospectively reviewed from the period between February 2000 and January 2017; the same cohort of patients was analysed in two other publications [Citation23,Citation24]. ACH is a tertiary public hospital and a clinical research facility that provides specialist leukaemia care to approximately 1.7 million people. Study procedures were approved by the Auckland District Health Board Research Review Committee (approval number A+7753). Patients were diagnosed based on the criteria specified by the World Health Organisation [Citation3]. The following main clinical and laboratory parameters were collected: age, gender, presenting full blood count, morphology, basic coagulation profile, molecular results, time between admission to our hospital and initiation of ATRA and chemotherapy treatment, and transfusion requirements during the first month. Outcomes of interest were early haemorrhagic and thrombotic complications, early deaths and long-term survival. Data were anonymised and analysed retrospectively.
The NZCR was queried to collect demographic data on APL patients diagnosed in all national centres for the period between January 2000 and December 2017. The NZCR has been collecting data on all malignant tumours diagnosed in New Zealand since 1948. The primary function of the registry is to provide information on the incidence and mortality from cancer. The registry follows guidelines for recording and reporting cancer incidence recommended by the International Agency for Research on Cancer and the International Association of Cancer Registries. Clinical laboratories are the primary source of data for the registry, with diagnostic information collected from pathology reports. Patient records are checked against hospital discharge information on the National Minimum Dataset. Ethnicity is assigned according to the Ministry of Health’s Ethnicity Data Protocols, where the respondent must identify their own ethnicity (self-identification). For the purpose of this study, APL patients recorded in the NZCR were identified based on the ICD-10 code C92.4. Data on ethnicity, age, gender, date of presentation and death were extracted on 14 April 2020, and analysed after de-identification.
2.2. Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics software version 25 and R version 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria). Data are presented as mean (standard deviation, SD) for parametric data, or median (interquartile range, IQR) for non-parametric data. Normality was assessed via Shapiro–Wilk test. Differences in categorical variables were compared using Pearson Chi-square test if n > 5 or Fisher’s exact test if n < 5. Mean differences of continuous variables were compared using two-sided, independent samples Student t-test for parametric data, or Mann–Whitney U test for non-parametric data. Survival curves were estimated using Kaplan-Meier analysis, and compared via the log-rank test. The date of censorship was 31 March 2018 for the Auckland cohort and 14 April 2020 for the NZCR cohort. A P < 0.05 was deemed statistically significant.
3. Results
3.1. Analysis of patients treated at Auckland City Hospital
3.1.1. Presenting characteristics
There were 70 patients with APL treated at ACH from February 2000 to January 2017. This report analysed 55 patients, 39 (71%) of European descent and 16 (29%) of Polynesian descent (10 Māori and 6 Pacific Islander patients). The remaining 15 patients were of other ethnicities, and were excluded from further analysis.
Diagnosis of APL was made using PML protein immunofluorescence test (PML IF; performed for all patients), karyotyping (performed for 52, 94.5% patients), fluorescence in situ hybridisation (FISH; performed for 40, 72.7% patients), and reverse transcription polymerase chain reaction (RT–PCR; performed for 42, 76.4% patients); each patient had at least one molecular test performed. All patients had t(15;17)(q24;q21) with the PML-RARA fusion detected by FISH and/or RT–PCR, no variant fusions were identified in this cohort. Of 42 patients in whom the PML breakpoint was documented, the most common was BCR1 (25, 59.5%), followed by BCR3 (16, 38.1%) and BCR2 (1, 2.4%). In addition to t(15;17)(q24;q21), 13 (23.6%) patients had other karyotypic changes, most commonly trisomy 8. There were no significant differences in the genetic findings between European and Polynesian patients ().
Table 1. Clinical and laboratory features of patients with acute promyelocytic leukaemia diagnosed at Auckland City Hospital from February 2000 to January 2017.
The hypergranular variant was the most common, with 10 (71.4%) NZ Polynesian and 30 (85.7%) NZ European patients having this morphology. The microgranular variant was twice as common in Polynesian patients (28.6%) compared to European (14.3%) but the difference did not reach statistical significance (P = 0.254; ).
The mean age of diagnosis for the whole cohort of APL patients managed at ACH was 46 years (median 47 years). Polynesian patients were diagnosed at a younger age (mean 34.2, median 33.5 years) compared with European patients (mean 50.5, median 54 years; P = 0.005; ). The frequency of APL increased with age, peaking in the sixth decade of life in European patients ((A–i)). In contrast, most Polynesian patients presented in their second and third decades of life. Eleven of 16 (68.8%) Polynesian patients were diagnosed before the age of 40 years, which compared to a much lower 10 of 39 (25.6%) in European patients (P = 0.005; (A–i)). The number of males and females in both Polynesian and European groups were roughly equal ().
Figure 1. Age distribution and survival of New Zealand Polynesian and European patients with acute promyelocytic leukaemia (APL). (A) Age at diagnosis of APL patients diagnosed at Auckland City Hospital (A–i) and recorded in the New Zealand Cancer Registry (A–ii) during 2000–2017. Bars indicate numbers of Polynesian patients (n = 16 in Auckland [i] and n = 46 in New Zealand [ii]) and European patients (n = 39 in Auckland [i] and n = 127 in New Zealand [ii]) categorized by their age at diagnosis. (B) Kaplan-Meier curves comparing cumulative survival probability between Polynesian and European patients with APL treated in Auckland City Hospital (B–i) and recorded in the New Zealand Cancer Registry (B–ii) from 2000–2017.
![Figure 1. Age distribution and survival of New Zealand Polynesian and European patients with acute promyelocytic leukaemia (APL). (A) Age at diagnosis of APL patients diagnosed at Auckland City Hospital (A–i) and recorded in the New Zealand Cancer Registry (A–ii) during 2000–2017. Bars indicate numbers of Polynesian patients (n = 16 in Auckland [i] and n = 46 in New Zealand [ii]) and European patients (n = 39 in Auckland [i] and n = 127 in New Zealand [ii]) categorized by their age at diagnosis. (B) Kaplan-Meier curves comparing cumulative survival probability between Polynesian and European patients with APL treated in Auckland City Hospital (B–i) and recorded in the New Zealand Cancer Registry (B–ii) from 2000–2017.](/cms/asset/0553d02d-14f8-4f5f-918d-b5cac62a51ef/yhem_a_1882146_f0001_oc.jpg)
Presenting blast counts were higher in Polynesians (2.2 × 109/L versus 0.04 × 109/L in Europeans; P = 0.033; ). Presenting total white cell count (WCC) also trended higher in Polynesian patients (4.3 × 109/L compared with 1.9 × 109/L in Europeans; P = 0.080). Activated partial thromboplastin time (APTT) and fibrinogen levels at presentation were not different between Polynesians and Europeans. However, prothrombin ratio was marginally higher in Polynesians (1.3 versus 1.2 in Europeans; P = 0.020). The remaining variables within the full blood count (haemoglobin level, platelet and neutrophil counts) were similar between Polynesian and European patients ().
The prognostic risk at presentation was assessed based on the criteria outlined by Sanz et al. [Citation25]. Of 16 Polynesian patients, 4 (25%) were high-risk (WCC > 10 × 109/L) and another 4 (25%) were low-risk (WCC ≤ 10 × 109/L, platelets > 40 × 109/L; ). In comparison, of 37 European patients for whom presenting counts were available, 6 (16.2%) were high-risk and 14 (37.8%) were low-risk. The higher percentage of high-risk patients amongst Polynesians appeared congruent with their higher blast counts, but this difference was not statistically significant (P = 0.630, ).
3.1.2. Treatment and outcomes
When compared to European patients, Polynesian patients had a shorter median time to ATRA initiation (5.4 h versus 16.2 h, calculated from the time of admission to our centre; P = 0.021; ). There was no difference in the length of time to commencement of chemotherapy (P = 0.907). Early outcomes (within the first 30 days) did not vary between Polynesian and European patients, with similar rates of thrombo-haemorrhagic complications and death in both groups, and similar transfusion requirements ().
Table 2. Time to treatment (ATRA and chemotherapy), bleeding/thrombotic events, and transfusion requirements during induction.
There were 16 deaths in the Auckland cohort, of which 5 (31.3%) were Polynesian and 11 (28.9%) European patients (P = 1.000; ). Of these, three patients (one Polynesian and two European) died within 30 days from diagnosis. Only two patients relapsed (both were European); one succumbed to APL, the other received haematopoietic stem cell transplant and is a long-term survivor. Long-term survival probability was similar between Polynesian and European patients (P = 0.600; (B–i)). Overall, there was no evidence that treatment outcomes differed between Polynesian and European patients.
Table 3. Long-term outcomes of patients with acute promyelocytic leukaemia treated at Auckland City Hospital.
3.2. Analysis of APL patients recorded in the New Zealand Cancer Registry
To verify demographic characteristics and long-term outcomes for Polynesian and European patients identified in the Auckland cohort, data was obtained from the NZCR for the period between January 2000 and December 2017. There were 198 patients with APL recorded in the NZCR over this time, including 127 (64.1%) of European descent and 46 (23.2%) of Polynesian descent (29 [14.6%] Māori and 17 [8.6%] Pacific Islander patients), all proportionate to the composition of New Zealand population. Four patients from the Auckland cohort were not captured by the NZCR (including two European and one Māori patients). Further analysis excluded 25 patients of other ethnicities.
Similar to the results obtained from ACH (), Polynesian patients with APL recorded in the NZCR were diagnosed at a significantly younger average age compared to European patients (35.6 versus 51.1 years respectively, P < 0.001; ; (A–ii)). The ratio of male to female in Polynesian and European groups were roughly the same. There were 60 deaths recorded during this time, of which 16 (34.8%) were Polynesian patients and 44 (34.6%) were European patients (P = 0.987; ). Long-term survival probability did not differ between Polynesian and European patients (P = 0.920; (B–ii)).
Table 4. Epidemiological data for patients with acute promyelocytic leukaemia recorded in the New Zealand Cancer Registry.
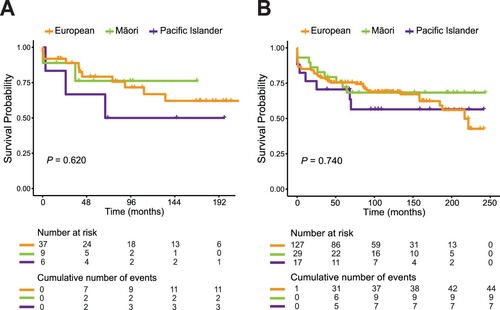
Finally, whilst we found certain distinctive features of APL in NZ Polynesian patients compared with European, there were no significant differences between Māori and Pacific Island patients for any of the parameters analysed, both from ACH (Supplementary Table S1) and NZCR (Supplementary Table S2). Long-term outcomes were similar between Māori and Pacific Island patients but will require confirmation in larger studies ().
Figure 2. Overall survival of European, Māori and Pacific Island patients with acute promyelocytic leukaemia (APL). Kaplan-Meier curves comparing cumulative survival probability between European, Māori and Pacific Island patients with APL treated in Auckland City Hospital (A) and recorded in the New Zealand Cancer Registry (B) between 2000 and 2017.
4. Discussion
This study highlights significant ethnic differences in the presenting features of APL between NZ Polynesian and European patients. We found that Polynesian patients presented at a younger age than European showing laboratory features suggestive of more severe disease. Reassuringly, Polynesian patients were started on ATRA faster and treatment outcomes were similar between both ethnic groups.
The median age of Polynesian patients with APL treated at ACH was 33.5 years and 35.0 years for patients recorded in the NZCR. In contrast, the median age of European patients was higher, 54.0 years at ACH and 52.0 years in the NZCR (P ≤ 0.005 for both). Median values from both sources were similar, supporting the generalisability of these results across New Zealand. Internationally, the median age of APL patients varies widely from 29 to 54 years [Citation26–32]. This variability may be a reflection of ethnic differences in the age of presentation as seen in this study. Many reports are from clinical trials that often have age restrictions, which may skew the true median age of patients in the population. Although the age of Polynesian patients was lower in our study compared with European patients, neither was obviously different from median ages reported worldwide.
NZ Polynesian patients had laboratory features suggestive of higher risk disease. Median circulating blast count was 2.2 × 109/L in Polynesians and 0.04 × 109/L in Europeans (P = 0.033); median WCC was 4.3 × 109/L in Polynesians and 1.9 × 109/L in Europeans (P = 0.080). A greater WCC in APL at presentation is known to associate with a higher risk of relapse [Citation25,Citation33]. Congruently, we found a higher percentage of high-risk patients [Citation25] among Polynesians compared to Europeans (25% and 16.2% respectively). Although the distribution of risk did not differ statistically (likely due to small numbers of patients), higher blast counts in Polynesians may be influencing higher risk, and further evaluation in larger cohorts is warranted.
WCC is often normal or low in hypergranular APL but usually elevated in microgranular APL (median count 1.8 and 15.8 × 109/L respectively) [Citation34]. Our results showed that microgranular variant correlated with higher WCC (data not shown) and was also more predominant amongst NZ Polynesians (28.6% versus 14.3% in Europeans, P = 0.254). This may suggest certain variants of APL are more common among different ethnicities, which may explain differences in the WCC and potentially, severity of APL between NZ Polynesian and European patients. Reminiscent of these findings, our previous research found that NZ Polynesian patients with polycythaemia vera (PV) also present at a younger age and with higher WCC [Citation35]. Together, these observations raise a possibility that NZ Polynesians are in some way predisposed to myeloid neoplasia, and further studies are warranted. While epidemiologic differences in haematologic malignancies have been comprehensively reviewed in recent years [Citation36,Citation37], further research is needed to better understand the underlying cause [Citation38]. Our findings suggest that differences in risk or genetic factors, more so than clinical management, underly variation in the severity of APL in NZ Polynesians. New Zealand indigenous population is relatively small and isolated, thus genetic linkage analysis may be easier compared to other, larger papulations such as Latino or Chinese.
The most frequent mutation that coincides with t(15;17)(q24;q21) is internal tandem duplication of the fms-like tyrosine kinase 3 gene (FLT3-ITD) [Citation39]. APL with FLT3-ITD mutation is associated with the microgranular variant, a higher WCC and higher numbers of circulating blasts [Citation40,Citation41]. It would be interesting to determine if NZ Polynesians have a higher frequency of the FLT3-ITD mutation or another specific genetic mutation that drives cell proliferation. Unfortunately, there are few genetic studies in NZ Māori and Pacific populations overall, and therefore understanding of genetic contributions to many diseases in this population is limited [Citation42]. Clinical significance of racially inclusive modern genomic testing at a population level in New Zealand is underscored by the discovery that a missense variant of CREBRF (CREB3 [cyclic AMP responsive element binding protein 3] regulatory factor; rs373863828-A) is common in NZ Polynesians but rare in non-Polynesian populations [Citation43–45]. This variant associates with higher body mass index (BMI); therefore its higher frequency in Polynesians may explain the greater prevalence of obesity in this group. Obesity is of relevance in APL epidemiology. Estey et al were the first to report the association between APL and obesity in a cohort of 1245 patients with AML (including 120 with APL) treated at the MD Anderson Cancer Center [Citation46]. In that cohort, increased BMI was strongly associated with the diagnosis of APL but not other AML (P = 0.0003) [Citation46]. Subsequent meta-analysis of 12,971 patients with AML (including 866 with APL) demonstrated that high BMI was associated with a higher incidence of all AML, but in contrast to non-APL, APL patients who were obese had worse outcomes, in particular shorter overall survival (P = 0.001), and a higher risk of differentiation syndrome (P = 0.040) [Citation47]. Considering NZ Polynesians have higher rates of obesity [Citation48], future studies should gather data for BMI to determine how it impacts APL presentation and outcomes in NZ Polynesian and European patients. Mechanistic examination of associated inflammatory pathways in this setting would be of interest [Citation49], including in the bone marrow microenvironment.
Racial disparities in treatment and outcomes of cancers are key issues for many minority and indigenous populations. In California, Black patients with AML had lower odds of receiving treatment compared to White patients [Citation50]. When the differences in dispensing of treatment were adjusted for, the hazard of mortality associated with Black ethnicity was significantly decreased, nearly completely eliminating the ethnic disparity [Citation50]; similar was recently reported for patients with plasma cell myeloma [Citation51]. Within the NZ population, racial disparities in treatment and outcomes have been identified in a number of solid cancer types, including prostate, breast, colon and lung cancers [Citation52]. In contrast, our study found that Polynesian patients had shorter median time interval to commencement of ATRA therapy when compared to European patients (5.4 h from admission versus 16.2 h respectively; P = 0.021), and times to chemotherapy dispensing were similar (62.1 h in Polynesians versus 50.0 h in Europeans; P = 0.907). Our results indicate a lack of racial disparity in the initiation of treatment for APL, and argue that Polynesian patients presented with more severe disease, that was appropriately managed with rapid emergency treatment. It is also possible that other concomitant conditions made Polynesian patients sicker at presentation, as rates of chronic conditions, such heart disease, renal failure and diabetes are higher amongst Polynesians [Citation53]. Our findings suggest that appropriate treatment of severe APL cases improved outcomes, as we found no difference in early adverse events and long-term survival between Polynesian and European patients. These are very reassuring findings for this patient group in our setting.
Our study should be considered in light of its limitations. This was a retrospective investigation involving relatively small numbers of patients, and treatment data was analysed from a single centre. However, to minimize the sampling bias associated with a small, single-centre cohort, results were corroborated with the national registry data where possible. Four patients from ACH were not captured by the NZCR, thus some information may be missing, but this is unavoidable. Limitations inherent to retrospective studies prevent any causative conclusions that can be made. We cannot explain what caused the difference in APL presentation between NZ Polynesian and European patients. Future studies are needed to investigate the underlying APL pathophysiology. Because ethnicity was self-reported, genetic testing would help identify links between genetic ancestry and disease risk. We hypothesize that NZ Polynesians carry increased genetic predisposition to APL that heightens cell proliferation, either directly or through indirect bone marrow niche effects. However, different risk factors and comorbidities may also be contributing, and these require evaluation in larger cohorts.
In conclusion, this study demonstrates key differences in the clinical presentation of NZ Polynesian and European patients with APL. Polynesian patients were younger at presentation and showed higher blast counts. However, appropriate treatment was administered quickly with no evidence of disparities in outcomes. A better understanding of the underlying cause for the phenotypic differences found may further our understanding of APL mechanisms, and suggest novel avenues for therapeutic research. We hope our evidence encourages further studies using novel genomic diagnostic and prognostic methods in vulnerable ethnic minorities internationally. Genetic differences between ethnic groups may play an under-appreciated role in differential susceptibility and outcomes of APL. Genome-wide association studies between NZ Polynesian and European patients with and without APL may identify new genetic drivers and cancer pathways amenable to novel preventative, diagnostic and therapeutic measures, ultimately leading to better outcomes for patients.
Author contributions
M.L.K-Z designed the study and N.C. collected clinica data; data analysis was done by C.V., V.Y.L., T.N.G. and M.L.K-Z; G.C. and E.T. provided supervision and mentoring. The paper was written by T.I., M.L.K-Z., C.V. and V.Y.L. All authors edited the manuscript and approved it for submission.
Supplemental Material
Download MS Word (17.8 KB)Acknowledgements
Chris Lewis (Analytical Services, New Zealand Ministry of Health) extracted data from the New Zealand Cancer Registry. The clinical haematology team at Auckland City Hospital provided outstanding care of APL patients. Ngā mihi nui.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;1:549–550. doi:https://doi.org/10.1016/s0140-6736(77)91415-5.
- Liquori A, Ibanez M, Sargas C, et al. Acute promyelocytic leukemia: a constellation of molecular events around a single PML-RARA fusion gene. Cancers (Basel). 2020;12;624. doi:https://doi.org/10.3390/cancers12030624.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi:https://doi.org/10.1182/blood-2009-03-209262.
- David S, Mathews V. Mechanisms and management of coagulopathy in acute promyelocytic leukemia. Thromb Res. 2018;164(Suppl 1):S82–S88. doi:https://doi.org/10.1016/j.thromres.2018.01.041.
- Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi:https://doi.org/10.1056/NEJMoa1300874.
- Douer D, Preston-Martin S, Chang E, et al. High frequency of acute promyelocytic leukemia among Latinos with acute myeloid leukemia. Blood. 1996;87:308–313. https://www.ncbi.nlm.nih.gov/pubmed/8547657.
- Douer D. The epidemiology of acute promyelocytic leukaemia. Best Pract Res Clin Haematol. 2003;16:357–367. doi:https://doi.org/10.1016/s1521-6926(03)00065-3.
- Rego EM, Jacomo RH. Epidemiology and treatment of acute promyelocytic leukemia in latin america. Mediterr J Hematol Infect Dis. 2011;3:e2011049, doi:https://doi.org/10.4084/MJHID.2011.049.
- Gómez-Almaguer D, Marcos-Ramírez ER, Montaño-Figueroa EH, et al. Acute leukemia characteristics are different around the world: the Mexican perspective. Clin Lymphoma Myeloma Leuk. 2017;17:46–51. doi:https://doi.org/10.1016/j.clml.2016.09.003.
- Douer D, Santillana S, Ramezani L, et al. Acute promyelocytic leukaemia in patients originating in Latin America is associated with an increased frequency of the bcr1 subtype of the PML/RARalpha fusion gene. Br J Haematol. 2003;122:563–570. doi:https://doi.org/10.1046/j.1365-2141.2003.04480.x.
- So CC, Wan TS, Chow JL, et al. A single-center cytogenetic study of 629 Chinese patients with de novo acute myeloid leukemia–evidence of major ethnic differences and a high prevalence of acute promyelocytic leukemia in Chinese patients. Cancer Genet. 2011;204:430–438. doi:https://doi.org/10.1016/j.cancergen.2011.06.003.
- Xu XJ, Tang YM, Song H, et al. Long-term outcome of childhood acute myeloid leukemia in a developing country: experience from a children’s hospital in China. Leuk Lymphoma. 2010;51:2262–2269. doi:https://doi.org/10.3109/10428194.2010.518653.
- Zhang L, Zhu X. Epidemiology, diagnosis and treatment of acute promyelocytic leukemia in children: the experience in China. Mediterr J Hematol Infect Dis. 2012;4:e2012012, doi:https://doi.org/10.4084/MJHID.2012.012.
- Testi AM, D'Angio M, Locatelli F, et al. Acute promyelocytic leukemia (APL): comparison between children and adults. Mediterr J Hematol Infect Dis. 2014;6:e2014032, doi:https://doi.org/10.4084/MJHID.2014.032.
- Summary of the New Zealand Population 1991-2018 Stats NZ. Available from: https://www.stats.govt.nz/topics/population.
- Soeberg M, Blakely T, Sarfati D, et al. Cancer Trends: Trends in cancer survival by ethnic and socioeconomic group, New Zealand 1991–2004. University of Otago and Ministry of Health (2012), https://www.health.govt.nz/publication/cancer-trends-trends-cancer-survival-ethnic-and-socioeconomic-group-new-zealand-1991-2004.
- Pacific People in NZ Ministry of Pacific Peoples. (2019). Available from: https://www.mpp.govt.nz/pacific-people-in-nz.
- Dachs GU, Currie MJ, McKenzie F, et al. Cancer disparities in indigenous Polynesian populations: Māori, Native Hawaiians, and Pacific people. Lancet Oncol. 2008;9:473–484.
- Teng AM, Atkinson J, Disney G, et al. Ethnic inequalities in cancer incidence and mortality: census-linked cohort studies with 87 million years of person-time follow-up. BMC Cancer. 2016;16:755, doi:https://doi.org/10.1186/s12885-016-2781-4.
- Dockerty JD, Cox B, Cockburn MG. Childhood leukaemias in New Zealand: time trends and ethnic differences. Br J Cancer. 1996;73:1141–1147. doi:https://doi.org/10.1038/bjc.1996.219.
- Tracey MC, Carter JM. Ethnicity variables in the incidence rates of leukemias in New Zealand populations: implications for stem-cell transplantation. Am J Hematol. 2005;79:114–118. doi:https://doi.org/10.1002/ajh.20355.
- Velickovic ZM, Carter JM. Feasibility of finding an unrelated bone marrow donor on international registries for New Zealand patients. Bone Marrow Transplant. 1999;23:291–294. doi:https://doi.org/10.1038/sj.bmt.1701561.
- Chien N, Petrasich M, Chan G, et al. Early treatment of acute promyelocytic leukaemia is accurately guided by the PML protein localisation pattern: real-life experience from a tertiary New Zealand centre. Pathology. 2019;51:412–420. doi:https://doi.org/10.1016/j.pathol.2019.01.003.
- Chien N, Varghese C, Green TN, et al. Treatment outcomes of patients with acute promyelocytic leukaemia between 2000 and 2017, a retrospective, single centre experience. Leuk Res. 2020;93:106358, doi:https://doi.org/10.1016/j.leukres.2020.106358.
- Sanz MA, Lo Coco F, Martin G, et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood. 2000;96:1247–1253. https://www.ncbi.nlm.nih.gov/pubmed/10942364.
- Muchtar E, Vidal L, Ram R, et al. The role of maintenance therapy in acute promyelocytic leukemia in the first complete remission. Cochrane Database Syst Rev (2013) CD009594. https://doi.org/https://doi.org/10.1002/14651858.CD009594.pub2.
- Iland HJ, Collins M, Bradstock K, et al. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: a non-randomised phase 2 trial. Lancet Haematol. 2015;2:e357–e366. doi:https://doi.org/10.1016/S2352-3026(15)00115-5.
- Jacomo RH, Melo RA, Souto FR, et al. Clinical features and outcomes of 134 Brazilians with acute promyelocytic leukemia who received ATRA and anthracyclines. Haematologica. 2007;92:1431–1432. doi:https://doi.org/10.3324/haematol.10874.
- Khorshid O, Diaa A, Moaty MA, et al. Clinical features and treatment outcome of acute promyelocytic leukemia patients treated at Cairo national cancer institute in Egypt. Mediterr J Hematol Infect Dis. 2011;3:e2011060, doi:https://doi.org/10.4084/MJHID.2011.060.
- Lehmann S, Ravn A, Carlsson L, et al. Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish adult acute leukemia Registry. Leukemia. 2011;25:1128–1134. doi:https://doi.org/10.1038/leu.2011.78.
- Pagoni M, Garofalaki M, Panitsas F, et al. Acute promyelocytic leukemia: an experience on 95 Greek patients treated in the all-trans-retinoic acid era. Mediterr J Hematol Infect Dis. 2011;3:e2011053, doi:https://doi.org/10.4084/MJHID.2011.053.
- Russell N, Burnett A, Hills R, et al. Attenuated arsenic trioxide plus ATRA therapy for newly diagnosed and relapsed APL: long-term follow-up of the AML17 trial. Blood. 2018;132:1452–1454. doi:https://doi.org/10.1182/blood-2018-05-851824.
- Daver N, Kantarjian H, Marcucci G, et al. Clinical characteristics and outcomes in patients with acute promyelocytic leukaemia and hyperleucocytosis. Br J Haematol. 2015;168:646–653. doi:https://doi.org/10.1111/bjh.13189.
- Tallman MS, Kim HT, Montesinos P, et al. Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA group. Blood. 2010;116:5650–5659. doi:https://doi.org/10.1182/blood-2010-06-288613.
- Hanna MZ, Kalev-Zylinska ML, Jackson SR, et al. Distinctive features of polycythaemia vera in New Zealand Polynesians. N Z Med J. 2018;131:38–45. https://www.ncbi.nlm.nih.gov/pubmed/30235191.
- Kirtane K, Lee SJ. Racial and ethnic disparities in hematologic malignancies. Blood. 2017;130:1699–1705. doi:https://doi.org/10.1182/blood-2017-04-778225.
- Bispo JAB, Pinheiro PS, Kobetz EK. Epidemiology and etiology of leukemia and lymphoma. Cold Spring Harb Perspect Med. 2020;10:a034819; doi:https://doi.org/10.1101/cshperspect.a034819.
- Landgren O. Racial/ethnic disparities: need more work!. Blood. 2017;130:1685–1686. doi:https://doi.org/10.1182/blood-2017-08-798546.
- Chen C, Huang X, Wang K, et al. Early mortality in acute promyelocytic leukemia: potential predictors. Oncol Lett. 2018;15:4061–4069. doi:https://doi.org/10.3892/ol.2018.7854.
- Marasca R, Maffei R, Zucchini P, et al. Gene expression profiling of acute promyelocytic leukaemia identifies two subtypes mainly associated with FLT3 mutational status. Leukemia. 2006;20:103–114. doi:https://doi.org/10.1038/sj.leu.2404000.
- Beitinjaneh A, Jang S, Roukoz H, et al. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: a systematic review. Leuk Res. 2010;34:831–836. doi:https://doi.org/10.1016/j.leukres.2010.01.001.
- Merriman TR, Wilcox PL. Cardio-metabolic disease genetic risk factors among Maori and Pacific Island people in Aotearoa New Zealand: current state of knowledge and future directions. Ann Hum Biol. 2018;45:202–214. doi:https://doi.org/10.1080/03014460.2018.1461929.
- Minster RL, Hawley NL, Su CT, et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat Genet. 2016;48:1049–1054. doi:https://doi.org/10.1038/ng.3620.
- Berry SD, Walker CG, Ly K, et al. Widespread prevalence of a CREBRF variant amongst Māori and Pacific children is associated with weight and height in early childhood. Int J Obes. 2018;42:603–607. doi:https://doi.org/10.1038/ijo.2017.230.
- Krishnan M, Major TJ, Topless RK, et al. Discordant association of the CREBRF rs373863828 A allele with increased BMI and protection from type 2 diabetes in Maori and Pacific (Polynesian) people living in Aotearoa/New Zealand. Diabetologia. 2018;61:1603–1613. doi:https://doi.org/10.1007/s00125-018-4623-1.
- Estey E, Thall P, Kantarjian H, et al. Association between increased body mass index and a diagnosis of acute promyelocytic leukemia in patients with acute myeloid leukemia. Leukemia. 1997;11:1661–1664. doi:https://doi.org/10.1038/sj.leu.2400783.
- Li S, Chen L, Jin W, et al. Influence of body mass index on incidence and prognosis of acute myeloid leukemia and acute promyelocytic leukemia: A meta-analysis. Sci Rep. 2017;7:17998, doi:https://doi.org/10.1038/s41598-017-18278-x.
- Duncan E, Schofield G, Duncan S, et al. Ethnicity and body fatness in New Zealanders. N Z Med J. 2004;117:U913, https://www.ncbi.nlm.nih.gov/pubmed/15282625.
- Deng T, Lyon CJ, Bergin S, et al. Obesity, inflammation, and cancer. Annu Rev Pathol. 2016;11:421–449. doi:https://doi.org/10.1146/annurev-pathol-012615-044359.
- Patel MI, Ma Y, Mitchell B, et al. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Epidemiol Biomarkers Prev. 2015;24:344, doi:https://doi.org/10.1158/1055-9965.EPI-14-0963.
- Fillmore NR, Yellapragada SV, Ifeorah C, et al. With equal access, African American patients have superior survival compared to white patients with multiple myeloma: a VA study. Blood. 2019;133:2615–2618. doi:https://doi.org/10.1182/blood.2019000406.
- Rahiri JL, Alexander Z, Harwood M, et al. Systematic review of disparities in surgical care for Maori in New Zealand. ANZ J Surg. 2018;88:683–689. doi:https://doi.org/10.1111/ans.14310.
- Blakely T, Ajwani S, Robson B, et al. Decades of disparity: widening ethnic mortality gaps from 1980 to 1999. N Z Med J. 2004;117:U995, https://www.ncbi.nlm.nih.gov/pubmed/15475978.