
ABSTRACT
Objectives
In this study, clinical and biochemical methods were utilized to predict the final diagnosis of hereditary spherocytosis (HS), correlate the diagnosis with splenectomy, and examine the usefulness of this approach.
Methods
We biochemically and cytochemically analysed erythrocyte membrane proteins before making a final HS diagnosis based on gene analysis to compare diagnostic approaches. The clinical features of six patients with various subtypes of HS and symptoms were observed by blood analysis using eosin-5′-maleimide staining, biochemical analysis using sodium dodecyl sulphate – polyacrylamide gel electrophoresis with western blotting, and mass spectrometry. Finally, diagnostic membrane gene analysis was performed.
Results
Five of the six patients showed mild to moderate or severe anaemia, and the other patient was non-anaemic; all six patients showed faint eosin-5′-maleimide staining. In western blotting of erythrocyte membrane proteins, all six patients (three with β-spectrin, two with ankyrin, and one with SLC4A1 anomalies) showed low-molecular-weight peptide fragments, which were confirmed by mass spectrometry in the region corresponding to the band 3 protein. The two patients with an ankyrin gene anomaly exhibited severe anaemia, and two patients with simultaneous SLC4A1, SPTB, and UGT1A1 anomalies exhibited mild anaemia and hyperbilirubinemia.
Discussion
We determined the relationship among clinical features, cytochemical parameters, and gene anomalies in six patients with newly diagnosed HS while referring to previously published cases.
Conclusion
These findings reveal a close relationship between clinical features and membrane characteristics in HS, which can facilitate diagnosis and inform treatment.
Introduction
Hereditary spherocytosis (HS) is the most prevalent congenital disease that causes haemolytic anaemia due to abnormalities in the erythrocyte membrane. In Japan, patients with HS account for 60% of all cases of congenital haemolytic anaemia [Citation1]. The erythrocyte membrane mainly consists of integral membrane proteins such as band 3 and a skeletal protein complex consisting of α- and β-spectrin, ankyrin, protein 4.1, protein 4.2, and actin. These membrane skeletal proteins interact with the integral membrane protein (band 3) to form an ankyrin – band 3 complex with spectrin tetramers, and form a junctional complex comprising actin, protein 4.2, protein 4.1, and band 3 (without ankyrin). Additionally, band 3 proteins exist in the membrane as a mixture of dimers and tetramers; dimers not linked to the ankyrin complex are known as free mobile band 3 [Citation2.]
Ankyrin abnormalities have been reported in 60% of patients with HS in western countries but in only 10% of patients with HS in Japan [Citation3]. Interestingly, in both western nations and Japan, the frequency of band 3 and spectrin abnormalities is approximately 20–30% [Citation3]. Genetic testing of genes associated with these major erythrocyte membrane proteins is required to diagnose the HS disease subtype [Citation4, Citation5]. The aim of this study was to improve diagnosis by utilizing clinical and biochemical methods to predict the final diagnosis of HS, correlate the diagnosis with splenectomy, and examine the usefulness of this approach.
Materials and methods
Patients and case history
The patients (three hospitalized patients and three out-patients) examined in this study were recruited for blood examination from our institute as well as from other institutes and hospitals. Four healthy adult controls were included for comparison. Written informed consent was obtained from all patients, and institutional review board approval was obtained for the study. The six patients examined are part of a cohort of 17 patients with HS who were part of a previous study on red blood cell membrane protein analysis [Citation6]. The four patients who provided informed consent for the genetic test were previously examined.[Citation4] The inclusion of a further two patients would likely yield more reliable results. These two patients and four healthy adults included controls provided informed consent.
The initial clinical findings of the six patients were collected according to the guidelines for the diagnosis and management of HS [Citation7], which are summarized in .
Table 1 Summary and comparison between clinical features, biochemical analysis and gene analysis in six patients with HS.
Controls four healthy adults (20–24 years old 2 female and 2 male) (C1)(C2)(C3)(C4)
Patient 1 (HS1) was a 15-year-old male without anaemia.
Patient 2 (HS2, reported as case H6 in the previous study [Citation6]) was a 22-year-old female with slight anaemia [haemoglobin (Hb) 11.8 g/dL at the time of blood and cytochemical testing]. At the age 6 years, she had a parvovirus B19 infection and underwent blood transfusion for severe anaemia (Hb 6.0 g/dL) and mild jaundice (indirect bilirubin, 4.2 mg/dL). The patient had also undergone gallbladder resection and splenectomy.
Patient 3 (HS3, reported as case HS3 in the previous study [Citation6]) was a 12-year-old male with slight anaemia (Hb 11.2 g/dL at the time of blood testing) with moderate jaundice (indirect bilirubin 5.0 mg/dL). Genetic analysis was performed at the age of 16 years.
Patient 4 (HS4) was a 6-year-old male with moderate anaemia. He had parvovirus B19 infection. Genetic analysis was performed at the age of 8 years, and he underwent splenectomy and gallbladder resection.
Patient 5 (HS5, reported as case HS1 in the previous study[Citation6]) was a 2.5-year-old male with severe anaemia. At birth, he underwent phototherapy and several blood transfusions. He suffered from parvovirus B19 infection at 18 months of age which caused the severe anaemia and made several blood transfusions necessary. He underwent splenectomy at 4 years of age.
Patient 6 (HS6, reported as case HS6 in the previous study [Citation6]) was a 2.5-year-old male with severe anaemia. At birth, he underwent phototherapy and several blood transfusions for severe anaemia (Hb 4.5 g/dL) and hyperbilirubinemia. He was diagnosed with parvovirus B19 infection with severe anaemia at 2 years of age. He underwent splenectomy and gallbladder resection at 4 years of age.
Analysis of erythrocyte membrane proteins
Haematological investigations were performed as described by Shibuya et al. [Citation6] Blood samples (2–3 mL) were collected from the patients with HS after obtaining informed consent. The peripheral blood samples containing red blood cells were washed with phosphate-buffered saline (pH 7.2) and labelled with eosin 5′-maleimide (EMA) at 4°C for 20 min. EMA-labelled band 3 protein on the extracellular side of the erythrocyte membrane was detected by confocal laser-scanning microscopy (FLUOVIEW FV10i, Olympus, Tokyo, Japan). The brightness was calculated for 30 red cells, and the values were averaged using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The control was set at 100%. A fluorescein isothiocyanate-conjugated anti-band 3 antibody (Santa Cruz Biotechnology, Dallas, TX, USA) was used to fluorescently label the intracellular domain of band 3 in isolated erythrocytes, fixed in 4% paraformaldehyde for 20 min, and then permeabilized with 0.3% Triton X at room temperature. The cells were treated with anti-band 3 monoclonal antibodies overnight; the staining pattern was visualized with fluorescein isothiocyanate-labelled secondary antibodies under confocal laser-scanning microscopy.
Erythrocyte membrane proteins were analysed using sodium dodecyl sulphate – polyacrylamide gel electrophoresis (SDS-PAGE), followed by Coomassie brilliant blue staining. The band sizes were measured using ImageJ software [Citation6].
Western blotting was performed to identify the fragmented low-molecular-weight membrane proteins (e.g. band 3, ankyrin, spectrin, and actin) using anti-band 3, anti-ankyrin, anti-spectrin (Santa Cruz Biotechnology), and anti-actin (Proteintech, Rosemont, IL, USA) antibodies, respectively.
Tryptic peptides from the band 3 region, obtained by SDS-PAGE, were analysed by matrix-assisted laser desorption ionisation-time-of-flight mass spectrometry (Bruker Co, Auto Flexll, Billerica, MA, USA) as reported previously [Citation7]. The sequence coverage (%) of the peptides was calculated using Mascot (Matrix Science. Co, Boston, MA, USA) search analysis software via the fingerprinting method [Citation8].
Finally, target capture sequence analysis of the erythrocyte membrane was performed by Professor Hitoshi Kanno (Tokyo Women’s Medical University).
Results
EMA binds to erythrocyte membranes; however, because of the lower number of target proteins, binding is typically weaker in patients with HS than in healthy individuals [Citation9]. In this study, EMA staining revealed moderate to dim brightness patterns in the six patients with HS compared to in the four healthy adults, who showed brilliant stained ( and (A)). Fluorescein isothiocyanate-conjugated anti-band 3 staining revealed thick, darkly stained intracellular bands in a ring-shaped pattern in all patients with HS, in contrast to the thin, ring-like pattern observed in the control samples ( and (B)).
Figure 1. (HS1–HS3) . (HS4–HS6). Erythrocyte membrane proteins in six patients with hereditary spherocytosis . (A) Eosin-5′-maleimide (EMA) staining (B) fluorescein isothiocyanate (FITC) staining of the intracellular domain of band 3. (C) Levels of band 3 protein in red blood cell membranes as determined by SDS-PAGE and Coomassie staining. (D) Densitometrical wave (E) MAS: mass spectrometry (spectrum patterns).
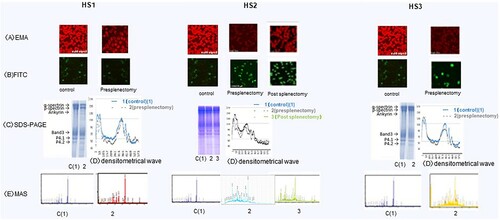
Figure 2. (HS1–HS3) . (HS4–HS6). Erythrocyte membrane proteins in six patients with hereditary spherocytosis. (A) Eosin-5′-maleimide (EMA) staining (B) fluorescein isothiocyanate (FITC) staining of the intracellular domain of band 3. (C) Levels of band 3 protein in red blood cell membranes as determined by SDS-PAGE and Coomassie staining. (D) Densitometrical wave (E) MAS: mass spectrometry (spectrum patterns).
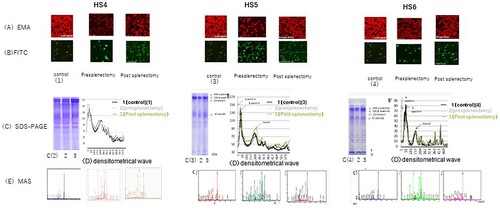
The erythrocyte membrane protein patterns and staining intensity of all patients with HS were similar to those of the controls after separation by SDS-PAGE ( and (C)), upper gel image), and showed similar levels based on the volume of the densitometrical waves ( and (D)) calculated using ImageJ software. Only patient HS4 exhibited a slightly lower staining intensity of band 3 ((C) (HS4)). Interactions between the low-molecular-mass, shorter-length proteins, and fragmented spectrin and ankyrin were observed by matrix-assisted laser desorption ionisation-time-of-flight mass spectrometry analysis. Peptides in the band 3 region (90–120 kDa) that were separated using SDS-PAGE in patients HS2–HS6, but not in patient HS1, were detected at an m/z 2000 ( and (E)); the sequence coverage results of the peptides are summarized in .
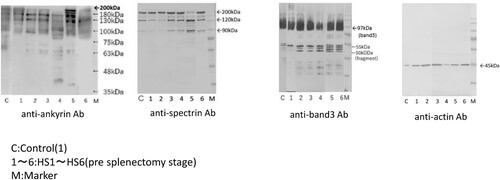
Western blotting in the controls and HS1–HS6 (pre-splenectomy stage) with anti-ankyrin revealed shorter-length ankyrin peptides with a lower estimated molecular mass (180, 130, and 94 kDa) compared to native ankyrin (approximately 200 kDa), along with several split low-volume ankyrin fragments (45–90 kDa) in patients HS2–HS6 (). Labelling with an anti-spectrin antibody revealed short-length spectrin proteins with lower molecular masses (120 and 90 kDa) than that of native spectrin (around 230–250 kDa) and degraded low-volume spectrin fragments (63–75 kDa) in patients HS2–HS6 () HS4–HS6). Several low-volume split band 3 fragments (50–55 kDa; native band 3 molecular mass is approximately 95 kDa) were observed in patients HS2–HS6; the number of these fragments was lower than that in the controls and in patient HS1-HS6 (). Furthermore, anti-actin antibodies detected a 45-kDa peptide in all six patients with HS and the controls (), which is larger than native actin (molecular mass of ∼42 kDa).
Figure 3. Western blotting (anti-spectrin Ab, anti-spectrin Ab, anti-band 3 Ab, anti-actin Ab).
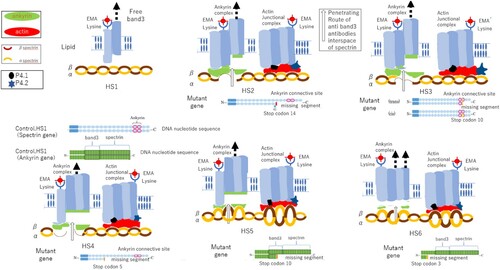
Patient erythrocyte membrane gene analysis revealed the following gene anomalies: solute carrier family 4 member 1 (SLC4A1) c.695-2a > c, (HS)inv-Exon19 splicing and bilirubin uridine diphosphate glucuronosyl transferase (UGT1A1) c.211G > A, p.G71R (Gilbert’s) heterozygote state in HS1; SPTB missense mutation c.5455G > T, p.E1819X heterozygote state in HS2; SPTB frameshift deletion c.4149-50insG, p.R1384AfsX7 heterozygote state in HS3; SPTB frameshift deletion c.2407>A, p.G71R heterozygote state and UGT1A1 c.211G > A, p. G71R (Gilbert’s) heterozygote state in HS4; ANK1 frameshift deletion c.1033 del G, p.A345fs441 heterozygote state in HS5; and ANK1 frameshift deletion c.382-6 del AACTT, p.K128F fsX7 heterozygote state in HS6 ().
The effects of these point mutations in the indicated erythrocyte membrane genes on the mechanism of release of band 3, spectrin, and ankyrin from the membrane are illustrated for each case in .
Discussion
We analysed six patients with HS showing abnormalities in the band 3 gene SLC4A1 (1 case), spectrin gene SPTB (3 cases), and ankyrin gene ANK1 (2 cases) through clinical and biochemical examinations. We observed a relationship among clinical features, cytochemical parameters, and gene anomalies. These data can help in predicting prognosis and selecting therapy.
One patient with an SPTB mutation showed a slight decrease in the levels of band 3 protein. This patient, along with two others, had an erythrocyte spectrin mutation resulting in dim staining by EMA, which was assumed to be because of the decreased binding force of the heterozygote mutant – deleted gene. The mutation introduces a stop codon in distal or proximal segment spectrins, causing one allele to be devoid of connective segments to ankyrin or band 3 and the other to produce spectrin proteins with weak and insufficient binding ability [Citation10], consequently, patient HS3 with the spectrin mutation showed low levels of band 3 protein ((C)HS3). This causes erythrocytes to become microspherocytes, resulting in a smaller amount of lysine residue on the extracellular band 3 of the erythrocyte membrane. In contrast, our results revealed abundant localization of the band 3 protein with thick and dark staining, possibly because splitting of spectrin and ankyrin results in many gap spaces in combination with spectrin, and the antibodies reach the intracellular domain band 3 ( and (HS1–6)).
Band 3 and protein 4.1, and protein 4.2 (as determined by SDS-PAGE, (C) and (C)), indicating that it remained in the erythrocyte membrane as a junctional complex [Citation11], therefore, not all band 3 proteins were released from the erythrocyte membrane ((HS2–6)). The actin band was verified via western blotting using an anti-actin antibody ().
Our patients with ankyrin gene mutation exhibited severe anaemia after birth and often undergo phototherapy to treat high bilirubinaemia as well as undergo frequent blood transfusions, as in the present cases (HS5 and HS6) [Citation12]. Furthermore, in western blotting and mass spectrometry, a low volume of ankyrin was determined to be the remaining protein in the membrane. This may be because these fragments are released from the erythrocyte membrane based on the heterozygote-mutant nature of the gene deletion along with the presence of a stop codon in the proximal segments. Thus, one allele is devoid of connective segments to spectrin and band 3, leading the other allele to produce proteins with only weak and insufficient binding ability to spectrin and band 3 ((HS5 and HS6)). In contrast, when the spectrin gene is mutated, if a stop codon appears in the distal segments, mild anaemia is typically observed but becomes more moderate when the stop codon appears proximally.
Figure 4. Mechanism of low-molecular-weight spectrin, and ankyrin production and loss of these protein complexes from the erythrocyte membrane due to specific gene anomalies observed in six patients with hereditary spherocytosis (HS1–HS6). HS1: Piece of band 3 released from the membrane because of a conformational change in intracellular band 3 during circulation in the spleen. HS2: Stop codon appears at 10 (distal area), and one allele missing seven segments forms the binding site and causes a lack of binding protein. HS3: Stop codon appears at 10 (distal area), and one allele missing seven segments forms the binding site and causes a lack of binding protein. HS4: Stop codon appears at 5 (proximal area) in the DNA sequence, and one allele missing 12 segments forms the binding site and causes a lack of binding protein. HS5: Stop codon appears at 10 (distal area) in the DNA sequence, and one allele missing 14 segments, comprising band 3 and spectrin, causes a lack of protein binding to half of band 3 and all spectrin. HS6: Stop codon appears at 3 (proximal area) in the DNA sequence, and one allele missing 21 segments, which comprises band 3 and spectrin, causes a lack of protein binding to all band 3 and spectrin.
Furthermore, we determined the usefulness of splenectomy in cases of patients who had undergone splenectomy by performing mass spectrometry analysis, which showed that the short-length spectrin and ankyrin peptide were reduced past the splenectomy stage. This may be because of the loss of the spleen, which is a destructive organ of red blood cells.
Finally, the patient with a mutation in SLC4A1 (which encodes band 3) was clinically diagnosed as non-anaemic, and biochemical analysis revealed no or very little low-molecular-weight band 3 remaining, with a slightly reduced amount of spectrin peptide detected by mass spectrometry only in the band 3 region. The erythrocytes were microspherocytic, and EMA staining was reduced; anaemia was not observed, and intracellular band 3 staining showed a thin ring-like pattern similar to in the controls. This was likely because the free band 3 dimer had not bound to spectrin and ankyrin. Alternatively, band 3 itself may have undergone a conformational change to become extracellular band 3 with a lysine residue buried in the membrane or was fragile with weak fixation ability to the membrane, thereby escaping from the erythrocyte membrane during circulation in the spleen. The absence of anaemia with moderate haemolysis (hyperbilirubinemia) in this case may have occurred because only a small part of extracellular band 3 was phagocytosed by splenic macrophages and was corrected by the production of red blood cells in the bone marrow. In this patient and in HS4, based on SPTB mutation analysis, hyperbilirubinemia was predicted to be due to moderate anaemia in the presence of the UGT1A1 (Gilbert’s) mutation, which is associated with congenital indirect hyperbilirubinemia and sometimes coexists with HS [Citation13].
Based on our findings, clinical observations along with laboratory and biochemical tests are useful for determining the HS disease subtype before genetic testing and splenectomy. A close relationship was found between the clinical features and membrane characteristics of HS, which will be useful for guiding splenectomy.
Recently, Andolfo I demonstrated the occurrence of multi-locus inheritance in 15% of patients diagnosed with RBC defect in volving piezo-1 which encodes PIEZO-1 and SPTA-1 variants [Citation14]. The other hand, Ma S reported the case of PIEZO-1 mutant (as known to mechanosensitive channel and force inducible conformational change protein) to congenital hemolytic anemia (named as to Xerocytosis or dehydrated hereditary stomatocytosis Type1 (DHS1) to splenectomy is contraindication) in African population [Citation15].
Furthermore, if PIEZO-1 and other mutant gene coexisting in cases with HS are accumulated, it will be contribute to the more precise identification of the subtype of HS and will be useful for selection of therapy.
Acknowledgements
We are grateful to Professor Hitoshi Kannno (Tokyo Women’s Medical University) for genetic analysis of hereditary spherocytosis membrane proteins. We also thank Dr Taroh Yamazaki of the Department of Paediatrics, Saitama Medical University for performing cytochemical tests for patients after obtaining informed consent. We would like to thank Editage (www.editage.com) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Yawata Y, Kanzaki A, Yawata A, et al. Hereditary red cell membrane disorders in Japan: their genotype and phenotype features in 1014 cases studied. Hematology 2001;6:399–422.
- Burton NM, Bruce LJ. Modelling the structure of the red cell membraneThis paper is one of a selection of papers published in a special issue entitled CSBMCB 53rd annual meeting — membrane proteins in health and disease, and has undergone the journal’s usual peer review process. Biochem Cell Biol 2011;89:200–215.
- Yawata Y, Kanzaki A, Yawata A, et al. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol 2000;71:118–135.
- Andolfo I, Russo R, Gambale A, et al. New insights on hereditary erythrocyte membrane defects. Haematologica 2016;101:1284–1294.
- Iolascon A, Androlfo I, Russo R. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haemotol 2019;187:13–24.
- Shibuya A, Kawashima H, Tanaka M. Analysis of erythrocyte membrane proteins in patients with hereditary spherocytosis and other types of hemolytic anaemia. Hematology 2018;23:669–615.
- Bolton-Maggs PHB, Langer JC, Iolascon A, et al. General haematology task force of the British committee for standards in haematology. guidelines for the diagnosis and management of hereditary spherocytosis–2011 update. Br J Haematol 2012;156:37–49.
- Aboleneen H, Wu H. Sequence, oligonucleotides blocked terminal using exonuclease digestion and electrospray mass spectrometry. Anal Biochem 2000;287:120–135.
- King MJ, Behrens J, Rogers C, et al. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated hemolytic anemia. Br J Haematol 2000;111:924–933.
- Hassoun H, Vassiliadis JN, Murray J, et al. Molecular basis of spectrin deficiency in beta spectrin Durham. A deletion within beta spectrin adjacent to the ankyrin binding site precludes spectrin attachment to the membrane in hereditary spherocytosis. J Clin Invest 1995;96:2623–2629.
- Lux SE, Palek J. Disorders of the red cell membrane. In: Hamdin RI, Lux SE, Stossel TF, editors. Blood principles and practice of hematology. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2000. p. 1709–1858.
- Satchwell TJ, Bell AJ, Hawley BR, et al. Severe ankyrin-R deficiency result in impaired surface retention and lysosomal degradational of RhAG in human erythroblasts. Haematologica 2016;101:1018–1027.
- Sharma S, Vukelja SJ, Kadakia S. Gilbert’s syndrome co-existing with and masking hereditary spherocytosis. Ann Hematol 1997;74:287–289.
- Andolfo I, Martone S, Rosato B E, et al. Complex Modes of Inheritance in Hereditary Red Blood Cell Disorders: A case Series Study of 155 Patients. Genes 2021;12:958–972.
- Ma S, Cahalon S, LaMonte G, et al. Common PIEZO-1 allele in African population causes RBC dehydration and attenuate plasmodium infection Cell. 2018;173:1–13.