
ABSTRACT
We describe a first Dutch case of Hb M Saskatoon (HBB:c.190C > T p.His64Tyr) in a 47-year-old female Dutch patient who presented with cyanosis, hemolysis, and abnormal co-oximetry. A mean corpuscular volume (MCV) of 105 fL caused by reticulocytosis (160 × 109/L) and low red blood cell count (3.6 × 1012/L) suggested an increased erythrocyte turnover. An HPLC glyco-globin analysis revealed a decreased HbA1c fraction of 12.3 mmol/mmol, HbA0 of 93.3% and an additional unidentified fraction at 1.2 min. DNA sequencing revealed a missense mutation in the HBB gene, (HBB:c.190C > T p.His64Tyr), known as Hb M Saskatoon, a variant which has been previously identified as an unstable hemoglobin variant leading to methemoglobinemia and anemia. In this report, we describe the clinical and remarkable laboratory aspects of our patient with Hb M Saskatoon, and the consequences for treatment and drug use.
Introduction
Human hemoglobin A (HbA) is a tetrameric protein consisting of two α-globins and two β-globins with noncovalently bound heme groups. Hemoglobinopathies are a group of genetic disorders resulting in quantitative or qualitative changes, leading to thalassemias or Hb-variants, respectively. Variants of the hemoglobin molecule may lead for example to altered functionality such as altered oxygen affinity, decreased stability, leading to thalassemia or chronic hemolysis in the case of hyper unstable Hb variants or methemoglobinemia. In case of the latter, the hemoglobin molecule is unable to bind oxygen due to the oxidized state of the iron ion in the heme molecule [Citation1,Citation2].
Clinical presentation and laboratory results
A 46-year-old Caucasian female patient with a medical history of psoriatic arthritis and irritable-bowel syndrome presented at the department of internal medicine of our hospital with a 3-month history of a debilitating fatigue and slowly progressive dyspnea on exertion. Vital signs were unremarkable although at home she had measured a saturation of 60% SpO2 on her pulse-oximeter. Physical examination revealed a distinct peripheral and central cyanosis which has been a pre-existent condition since her childhood (). She indicated that her father had experienced similar symptoms throughout his life. He had told her that he had been diagnosed with a hemoglobinopathy, but no medical information was available. Results of the laboratory analysis are summarized in . The analysis indicated macrocytosis, increased erythrocyte turnover and decreased HbA1c of 12.3 mmol/mol with a normal fraction of HbA0 (93.3%). A microscopic analysis of the red blood cell morphology showed no aberrations. The hemoglobin separation using a Tosoh HLC-723 G8 Glycohemoglobin Analyzer (Tosoh Bioscience, Inc., South San Francisco, CA) demonstrated an unidentified Hb fraction with an approximate retention time 1.15 min (, red arrow) that could be attributed to an unstable Hb variant. Sanger sequencing for HBA1, HBA2, and HBB was performed using the Big Dye Terminator kit v3.1 cycle sequencing (Applied Biosystems Inc., Foster City, CA, USA) and analysis was done on the ABI3730 according to the manufacturer’s instructions. The primers and conditions for amplification and sequencing of the HBA1, HBA2 and HBB gene were as described previously [Citation3]. The sequence analysis revealed heterozygosity for a variant in the HBB gene: HBB: c.190C > T, p.His64Tyr. In this variant, the histidine residue 64 of the beta-globin chain distal to the porphyrin cofactor has been substituted by a tyrosine, previously annotated as Hb M Saskatoon [Citation4]. This Hb variant has previously been described as biochemically unstable and prone to disintegration into dimers. In other case reports, a similar small peak in the chromatogram has been observed and assigned to methemoglobin [Citation5,Citation6].
Figure 1. Patient presentation. Noticeable are the cyanotic features in her extremities as well as distinct cyanotic discoloration of her lips.
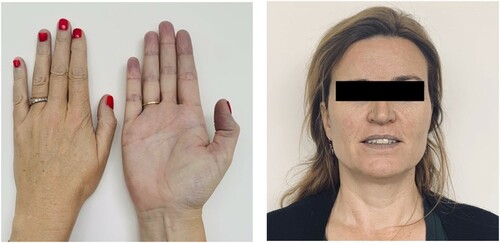
Figure 2. Chromatogram of the patient’s sample analyzed with a Tosoh HLC-723 G8 analyser using the standard HbA1c mode. HbA1c, in red, measured at 3.3% in this mode (12.3 mmol/mol) at 0.6 min. An additional unidentifiable peak is shown at approximately 1.15 min (arrow), indicating the presence of abnormal unstable Hb M Saskatoon.
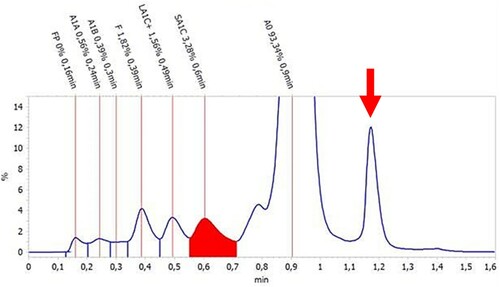
Table 1. Clinical chemistry and hematological parameters of the patient.
The genetic variant of Hb M Saskatoon was first reported in 1950 by Baltzan et al. and was found in a Canadian patient living in Saskatoon [Citation4]. Over decades, patients were identified with similar clinical symptoms and abnormal hemoglobin variants. As sequencing was not yet available for most of the last century, a number of hemoglobin variants were named after the geographical area or place of residence of the patient the variant was discovered in, but later turned out to be identical to Hb M Saskatoon. A summary of Hb M Saskatoon cases is shown in .
Table 2. Overview of case reports of patients presenting with HbM Saskatoon.
Hb M Saskatoon is a pathogenic variant leading to methemoglobinemia. Under physiological circumstances, a Fe2+ ion is embedded in a porphyrin covalently bound to the α- and β-globin chains of the hemoglobin protein, where it is able to bind to an oxygen molecule. Structural changes in the Hb M variants have the effect of stabilizing the heme iron atoms in the ferric (Fe3+) state, which makes the heme incapable of binding oxygen. In Hb M Saskatoon the mutation of histidine by a tyrosine residue compromises the hydrophobicity of the heme pocket and allows oxidation of the Fe2+ to the ferric (Fe3+) state, causing methemoglobinemia.
Due to the cyanotic appearance and indication of low oxygen saturation by a portable finger pulse-oximeter (Beurer), methemoglobin analysis was performed. The blood gas analyser (Siemens Rapidpoint500) indicated abnormal spectral absorbance and issued a warning for methemoglobin. Quantitation of methemoglobin could not be performed. Co-oximetry analysis was also performed using a second blood gas analyser (Radiometer ABL90 Flex Plus). This instrument also could not detect oxygen saturation or methemoglobin values, because of interference attributed to the presence of unstable methemoglobinemia. In a previous case of a patient carrying the Hb M Hyde Park variant, in which the histidine residue at position 92 of the β-globin chain has been substituted by a tyrosine residue, which is at the opposite (proximal) side of the porphyrin ring compared to the Hb M Saskatoon, methemoglobin also could not be detected [Citation23]. This observation indicates that unstable Hb M variants interfere with absorption spectra of stable variants in blood gas co-oxymetry, disabling the measurements of the of stable hemoglobins, like oxyHb (OHb), deoxyHb (HHb), carboxy Hb (COHb) and stable methemoglobin (metHb) [Citation24].
Methemoglobinemia
Methemoglobinemia may arise as a result of genetic or acquired causes. Acquired methemoglobinemia is most prevalent and commonly caused by oxidative stress, which can have exogenous or endogenous origins. Endogenous forms of oxidative stress include but are not limited to free radicals such as nitric oxide, hydrogen peroxide and hydroxyl radicals. Exogenous substances may also trigger the oxidation of hemoglobin and this occurs most often as a result of medication. Dapsone, topical anesthetics (benzocaine, lidocaine), anti-malarial agents (chloroquine, hydroxychloroquine) and street drugs account for the majority of iatrogenic methemoglobinemia [Citation25,Citation26]. In addition to the formation of methemoglobin in healthy patients, these drugs may seriously exacerbate the condition of patients who are prone to methemoglobinemia due to genetic mutations causing hemoglobinopathies. Genetic defects that result in methemoglobinemia are commonly found in the CYB5R3 gene, which encodes the cytochrome b5 reductase enzyme. This enzyme is responsible for the reduction of the Fe3+ ion in methemoglobin to Fe2+. Mutations in this gene can both affect the enzymatic stability as well as its catalytic activity [Citation27,Citation28]. Most other genetic defects giving rise to (unstable) methemoglobinemia are hemoglobinopathies that result in the formation of HbM variants due to the defects in the alpha, beta, or gamma globins [Citation29].
Precautions and treatment options
Methemoglobinemia can potentially be life-threatening, but only patients with methemoglobin levels >30% or >20% accompanied with symptoms require treatment. Treatment at lower levels should be considered in patients presenting with concomitant anemia or with a known cardiorespiratory medical history [Citation30]. The therapy depends largely on the cause of methemoglobinemia. In acquired methemoglobinemia, besides cessation of the causative agent and supportive therapy, methylene blue (MB) is preferred as treatment and previous reports have shown a rapid effect when administered in subsequent doses of 1–2 mg/kg intravenously up to a maximum of 7 mg/kg over 24 hours. However, MB should be administered with caution, since its effect is dependent on glucose-6-phophate dehydrogenase (G6PD) and NADH-MetHb-reductase. G6PD delivers nicotinamide adenine dinucleotide phosphate (NADPH) needed to convert MB to the metabolic product leukomethylene blue, which acts as a reducing agent to convert methemoglobin to hemoglobin. Therefore, MB is contraindicated in patients with G6PD deficiency or proven NADH-MetHb-reductase deficiency.
Alternatively, ascorbic acid acts as an antioxidant and has also shown to be effective in decreasing the level of methemoglobin. Ascorbic acid appears to act slower than MB taking up to 1–3 days as it often requires repetitive dosing of 2–40 g administered intravenously [Citation31]. Therefore, MB is usually preferred over ascorbic acid in acute methemoglobinemia although no randomized controlled trials have been performed to compare both treatments.
In the case of severe refractory methemoglobinemia, treatment with exchange transfusions and hyperbaric oxygen has shown improvement. However, little evidence exists concerning these two modalities which therefore remain a last resort in severe and refractory methemoglobinemia [Citation32,Citation33].
In Hb M disease, treatment with both MB and ascorbic acid is ineffective because the ferric state in Hb M is stabilized. The Hb M structural variants are unstable or characterized by decreased oxygen affinities rather than other causes of methemoglobinemia, such as oxidative stress or enzymatic deficiency (CYB5R3). Treatment with MB would be undesirable, because as an oxidating agent, MB itself poses a risk of hemolytic anemia and could exacerbate methemoglobinemia. Exposure to any oxidizing agent should be avoided since these patients exhibit an increased risk of progression to symptomatic methemoglobinemia. Since there are no effective treatment options for patients with Hb M disease, counseling should focus on reassuring the patient about their benign condition and offering genetic testing to first-degree relatives.
The patient in our case was prescribed oral folic acid 0.5 mg and vitamin B-12 1000 µg daily. Two months later, our patient reported increased fitness. Vitamin B12 and folic acid levels were measured 6 months after the start of oral supplements and measured at 83 nmol/L folic acid and 500 pmol/L vitamin B12. We hypothesize that an increased erythrocyte turnover in this patient with Hb M disease in combination with a relatively low level of serum folate and vitamin B-12 may have triggered a symptomatic anemia with a relative increase of dysfunctional Hb M.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Giardine BM, Joly P, Pissard S, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2021 Jan 8;49(D1):D1192–D1196.
- Hardison RC, Chui DH, Giardine B, et al. Hbvar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum Mutat. 2002 Mar;19(3):225–233.
- Harteveld CL. State of the art and new developments in molecular diagnostics for hemoglobinopathies in multiethnic societies. Int J Lab Hematol. 2014 Feb;36(1):1–12.
- Baltzan DM, Sugarman H. Hereditary cyanosis. Can Med Assoc J. 1950 Apr;62(4):348–350.
- Baine RM, Wright JM, Johnson MH, et al. Biosynthetic evidence for instability of HB M Saskatoon. Hemoglobin. 1980;4(2):201–207.
- García-Morin M, Manrique-Martin G, Ropero P, et al. Hb M-Saskatoon: an unusual cause of cyanosis in a Spanish child. Pediatric Hematology Oncology Journal. 2019;4(1):23–26.
- Shibata S, Miyaji T, Iuchi I, et al. Hemoglobin M-Kurume: its identity with hemoglobin M-Saskatoon. Nippon Ketsueki Gakkai Zasshi. 1962 Oct;25:690–694.
- Pik C, Raine DN. The chemical identification of two cases of Hb M (Hb M Boston and Hb M Saskatoon) occurring in England. Clin Chim Acta. 1964 Jul;10:90–92.
- Betke K, Kleihauer E, Gehring-Muller R, et al. [HbM Hamburg, a beta chain anomaly: alpha-2-beta-2-63Tyr (equals HbM Saskatoon)]. Klin Wochenschr. 1966 Aug 15;44(16):961–966.
- Hayashi A, Shimizu A, Yamamura Y, et al. Hemoglobins M: identification of Iwate, Boston, and Saskatoon variants. Science. 1966 Apr 8;152(3719):207–208.
- Efremov GD, Huisman TH, Stanulovic M, et al. Haemoglobin M Saskatoon and haemoglobin M Hyde Park in two Yugoslavian families. Scand J Haematol. 1974;13(1):48–60.
- Kohne E, Grosse HP, Versmold H, et al. [HbM Erlangen: alpha2beta263(e7) tyr. New mutation with haemolysis and NADH-methaemoglobin reductase deficiency (author's transl)]. Z Kinderheilkd. 1975 Jul 1;120(1):69–78.
- Molchanova TP, Abaturov LV, Spivak VA, et al. [Hemoglobin M Saskatoon alpha 2 beta 2 63 (E7) His–Tyr. Structural identification, hemichrome formation and proteolytic degradation]. Mol Biol (Mosk). 1980 Nov–Dec;14(6):1253–1266.
- Arbane-Dahmane M, Rouabhi F, Hocine M, et al. Hemoglobin M Saskatoon (alpha 2 beta 2 63(E7) His–>Tyr) in an Algerian family. Hemoglobin. 1985;9(5):509–511.
- Kazanets EG, Andreeva AP, Khangulov SV, et al. [Hereditary cyanosis caused by the presence of abnormal hemoglobin M in the blood: its detection, identification and properties]. Gematol Transfuziol. 1990 Mar;35(3):9–13.
- Waye JS, Patterson M, Eng B. De novo beta-globin gene mutation [beta 63(E7)His–>Tyr] giving rise to Hb M disease in a Newfoundlander. Hemoglobin. 1994 Nov;18(6):441–443.
- Suryantoro P, Takeshima Y, Haryanto A, et al. C to T transition at the first nucleotide of codon 63 of the beta-globin gene corresponding to hemoglobin M-Saskatoon in an Indonesian boy. Jpn J Hum Genet. 1995 Jun;40(2):195–201.
- Kedar PS, Nadkarni AH, Phanasgoankar S, et al. Congenital methemoglobinemia caused by Hb-MRatnagiri (beta-63CAT–>TAT, His–>Tyr) in an Indian family. Am J Hematol. 2005 Jun;79(2):168–170.
- Hutten M, Kohne E, Yagmur E, et al. Persistent cyanosis in a 4 month old infant with severe pneumonia and haemoglobin M. Klin Padiatr. 2009 Sep;221(5):305–307.
- Brunner-Agten S, Hergersberg M, Herklotz R, et al. Compound heterozygosity of Hb Hamilton and de novo mutated HbM Saskatoon. Ann Hematol. 2010 May;89(5):517–518.
- Akar N, Arslan C, Kurekci E. First observation of hemoglobin m Saskatoon (ss63 (e7) his > tyr(c-t)) in the iraqi population. Turk J Haematol. 2012 Sep;29(3):287–288.
- Gupta O, Chhabra S, Kaur J, et al. Unusual C- and A2-window peaks (hemoglobin M-Saskatoon) in three north Indian patients. Indian J Hematol Blood Transfus. 2019 Jan;35(1):196–198.
- Schiemsky T, Penders J, Kieffer D. Failing blood gas measurement due to methemoglobin forming hemoglobin variants: a case report and review of the literature. Acta Clin Belg. 2016 Jun;71(3):167–170.
- Zhao Y, Qiu L, Sun Y, et al. Optimal hemoglobin extinction coefficient data set for near-infrared spectroscopy. Biomed Opt Express. 2017 Nov 1;8(11):5151–5159.
- Coleman MD, Coleman NA. Drug-induced methaemoglobinaemia. Treatment issues. Drug Saf. 1996 Jun;14(6):394–405.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobinemia as a complication of topical dapsone. N Engl J Med. 2015 Jan 29;372(5):491–492.
- Katsube T, Sakamoto N, Kobayashi Y, et al. Exonic point mutations in NADH-cytochrome B5 reductase genes of homozygotes for hereditary methemoglobinemia, types I and III: putative mechanisms of tissue-dependent enzyme deficiency. Am J Hum Genet. 1991 Apr;48(4):799–808.
- Shirabe K, Yubisui T, Borgese N, et al. Enzymatic instability of NADH-cytochrome b5 reductase as a cause of hereditary methemoglobinemia type I (red cell type). J Biol Chem. 1992 Oct 5;267(28):20416–20421.
- Percy MJ, McFerran NV, Lappin TR. Disorders of oxidised haemoglobin. Blood Rev.. 2005 Mar;19(2):61–68.
- Murray L. Goldfrank’s toxicologic emergencies, 7th edition. Emerg Med Australas. 2004 Feb;16(1):87.
- Rehman A, Shehadeh M, Khirfan D, et al. Severe acute haemolytic anaemia associated with severe methaemoglobinaemia in a G6PD-deficient man. BMJ Case Rep. 2018 Mar 28. doi:https://doi.org/10.1136/bcr-2017-223369.
- Goldstein GM, Doull J. Treatment of nitrite-induced methemoglobinemia with hyperbaric oxygen. Proc Soc Exp Biol Med. 1971 Oct;138(1):137–139.
- Singh P, Rakesh K, Agarwal R, et al. Therapeutic whole blood exchange in the management of methaemoglobinemia: case series and systematic review of literature. Transfus Med. 2020 Jun;30(3):231–239.