
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Introduction
Allogeneic hematopoietic stem cell transplant (HSCT) and gene therapy (GT) are two potentially curative approaches for sickle cell disease (SCD), but they have never been compared in clinical trials.
Objective
To compare the safety and efficacy of HSCT and GT to assist clinicians and patients in making informed treatment decisions.
Methods
Phase I-III clinical trials and case reports/series were included. Regimens included HSCT from all stem cell sources, lentiviral gene therapy, and gene editing, with any conditioning regimen. We searched Medline and EMBASE databases as of 1st June 2020 for studies reporting HSCT and GT outcomes in SCD. The Newcastle-Ottawa scale was used to assess the risk of bias. Descriptive statistics and post-hoc imputation for standard deviations of mean change in FEV1 and FVC were performed.
Results
In total, 56 studies (HSCT, n = 53; GT, n = 3) representing 1,198 patients met inclusion criteria (HSCT, n = 1,158; GT, n = 40). Length of follow-up was 3,881.5 and 58.7 patient-years for HSCT and GT, respectively. Overall quality of evidence was low, with no randomized controlled trials identified. Two-year overall survival for HSCT was 91%; mortality was 2.5% for GT. Acute chest syndrome and vaso-occlusive episodes were reduced post-HSCT and GT. Meta-analysis was not possible due to lack of comparator and heterogeneity in outcome measures reporting. Very few studies reported post-transplant end-organ function. Six secondary malignancies (5 post-HSCT, 1 post-GT) were reported.
Discussion
Reporting of SCD-related complications and patient-important outcomes is lacking for both strategies. We advocate for standardized reporting to better compare outcomes within and between treatment groups.
Introduction
Sickle cell disease (SCD) results from a missense mutation in the β-globin gene (HBB), forming sickle hemoglobin (α2βS2; HbS), which self-polymerizes under prolonged deoxygenated conditions. Hemolysis and vaso-occlusion ensue, and manifest clinically as lifelong recurrent painful vaso-occlusive episodes (VOE) and cumulative organ damage. Despite interventions such as hydroxyurea, transfusion, newborn screening, vaccination, and antibiotic prophylaxis, disease morbidity and mortality remain unacceptably high, and overall survival (OS) in this patient population continues to lag behind that of the general population by approximately two decades, even in high-resource settings [Citation1,Citation2].
Both allogeneic hematopoietic stem cell transplant (HSCT) and gene therapy (GT) have emerged as potentially curative strategies for SCD over the last several decades. With HSCT, patients receive hematopoietic stem cells (HSCs) from HLA-matched related donors (MRDs), unrelated donors (MUDs), HLA-mismatched unrelated donors (MMUDs), umbilical, or haploidentical donors following a myeloablative, reduced-intensity, or non-myeloablative conditioning regimen [Citation3]. Successful engraftment of donor HSCs leads to long-term production of normal adult hemoglobin (α2β2, HbA). HSCT from MRDs is now employed by many centers in the treatment of patients with severe SCD, but is not without significant risks: conditioning regimen toxicity, graft failure, graft-versus-host disease (GVHD), and death are all potential consequences [Citation4].
GT is a newer strategy utilizing autologous HSCs. With this approach, ex vivo gene addition or editing techniques are used to reverse the SCD phenotype by inducing expression of anti-sickling β-globin or γ-globin (fetal globin) via silencing or disruption of the fetal globin repressor, BCL11A [Citation5,Citation6]. Current gene addition approaches use lentiviral vectors for ex vivo transduction of patient-derived HSCs with anti-sickling globin genes [Citation5]. Gene editing techniques use endonucleases to induce DNA double-stranded breaks at targeted sites and take advantage of endogenous non-homologous end joining to create insertions or deletions that disrupt the function of the targeted gene (e.g. BCL11A) [Citation5]. Following a myeloablative conditioning regimen similar to that used for HSCT, successful engraftment of modified patient-derived HSCs results in the expression of anti-sickling or fetal globin genes. While gene therapy still carries the risk of conditioning regimen-associated toxicity, the use of autologous over allogeneic HSCs eliminates GVHD risk and the need for HSC donors [Citation3].
Both HSCT and GT have demonstrated efficacy against SCD in clinical trials, yet these strategies have never been compared. The objective of this systematic review was to compare the efficacy and safety between HSCT and GT in patients with severe [Citation7] SCD.
Methods
This systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [Citation8]. Please see the Supporting Information for the study protocol and PRISMA checklist. The study protocol was not registered. Studies were included if they were Phase I-III clinical trials, case reports or series involving >1 patient at any age, undergoing HSCT or GT for cure of their SCD. The following potentially curative regimens were included: allogeneic stem cell transplant from all HSC sources, lentiviral gene therapy, and gene editing with targeted endonucleases. All types of conditioning regimens were included. Studies involving HSCT or GT for both SCD and β-thalassemia were included if results were reported separately for SCD patients. Non-English studies and preclinical studies examining the efficacy and safety of gene transfection or stem cell quality without infusion into the patient were excluded.
Primary outcomes were OS, event-free survival (EFS), graft failure, transplant-related mortality (TRM), time to neutrophil and platelet engraftment, and GVHD, and were defined per the European Society for Blood and Marrow Transplantation [Citation9] and Kanter et al. [Citation10]. An a priori decision was made to not analyze these outcomes based on transplant regimen and donor source given that this is not the objective of the study, and prior literature has reviewed this in detail [Citation11]. Donor chimerism was included as a primary outcome post-hoc. Secondary outcomes were secondary malignancies, occurrence of SCD complications post-transplant (acute chest syndrome (ACS), VOE, cerebrovascular accidents (CVA), avascular necrosis (AVN), retinopathy, and leg ulcers), end-organ function, health-related quality of life (HRQOL), opioid use, and hospitalization.
The literature search was performed using the Medline and EMBASE databases (see for search terms) on 11 July 2019 and an updated search on 1 June 2020. Both full-length articles and conference abstracts were included. References of included studies were screened for additional relevant studies. All titles were screened by two authors (L.E.R. & C.J.P.) for full text retrieval. The authors independently reviewed all retrieved full-text articles for inclusion in the review based on the predetermined criteria stated above. Agreement between the two authors was calculated using the kappa statistic. Disagreements were resolved by consensus between the two authors. One author (L.E.R.) extracted data on study characteristics, quality, and effects, and organized these data into table-form. Variables collected, in addition to the primary and secondary outcomes, included age and HSC donor type. The characteristics, interventions and outcomes of the included studies were summarized (see Tables S1 and S2). Settings and characteristics of the included studies did not influence how they were grouped within each comparison and were not defined a priori. All available outcomes from included studies were tabulated (see Tables S1 and S2). The definition of several outcomes (OS, EFS, chimerism, and time to platelet engraftment) varied across studies, and as such, these definitions were further restricted to facilitate synthesis (see Results section for definitions and results of the synthesis).
Table 1. Search terms used for Medline and EMBASE databases.
Descriptive statistics (proportion, mean and standard deviation, or median and range where applicable) were presented using Microsoft Excel, Version 16.43 (Microsoft, https://www.microsoft.com). All results associated with each outcome domain were reported with the exception of chimerism, where only 1-year chimerism was reported, and platelet engraftment, which was reported as >50 × 109/L.
We performed imputation statistics post-hoc for standard deviations of mean change in FEV1 and FVC where mean baseline and post-HSCT data were available, by first imputing the correlation coefficient according to the formula: [Citation12] using data from Walters et al. [Citation13]. For Tables 3 and 4, 95% confidence intervals were converted to standard deviations using the formula
in Microsoft Excel [Citation14]. Meta-analysis was planned (see Supporting Information for the study protocol), but could not be performed due to non-randomized nature, lack of comparator, and heterogeneity between studies. Risk of bias for all studies was appraised independently by two authors using the Newcastle-Ottawa Scale (NOS) [Citation15], which assesses quality of evidence of non-randomized studies. Authors assigned a score out of 4 for participant selection, a score out of 2 for comparability between groups, and a score out of 3 for reporting of outcomes, with higher scores indicating lower risk of bias. Any disagreements were resolved by consensus.
Results
Database search
We identified 949 titles through database searching on 11 July 2019 and an updated search on June 1, 2020 (see for PRISMA diagram). An additional 14 titles were identified from references of included studies, and 1 title was identified during peer review. After removal of duplicate citations (n = 147), exclusion of records not meeting eligibility criteria (n = 716, of which 60 were excluded following full-text review (Table S3)), and merging 45 studies with their primary citation, 56 unique studies were included for data extraction (). Agreement between the two reviewers was 90% and Cohen's kappa was 0.71. No randomized controlled trials were identified: all included studies were observational studies, retrospective chart reviews, or case reports/series. Only 2 included studies had a comparator group [Citation16,Citation17], comparing outcomes between transplanted patients and patients receiving standard medical therapy, but not between HSCT and GT.
Figure 1. PRISMA flow diagram of search results and inclusion and exclusion of citations.
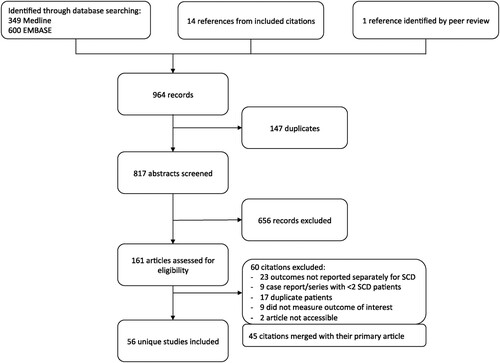
Table 2. Included studies and their risk of bias grading, using the Newcastle-Ottawa Scale.
Included studies
In total, 56 studies (53 HSCT, 3 GT) representing 1,198 (1,158 HSCT, 40 GT) patients were included. The median age of study participants ranged from 3.6–36 years. Of the included HSCT studies that reported participant age ranges, 14 studies (n = 304) reported outcomes in children only (<18 years of age) [Citation13,Citation16,Citation18–30], 6 studies (n = 57) reported outcomes in adults (≥18 years) [Citation31–36], and 29 studies (n = 624) transplanted children and adults [Citation37–65]. In the GT group, 2 of 3 studies reported outcomes in both adults and children (age range 12–38) [Citation66,Citation67], while the remaining study reported outcomes for 3 adults aged 21–26 [Citation68]. Donor hematopoietic stem cell source, reported for 1,032 HSCT patients, was MRD (n = 821), MUD (n = 73), haploidentical (n = 127), and MMUD (n = 11). GT strategy was lentiviral transduction with the Lentiglobin BB305 vector encoding the anti-sickling beta-A-T87Q globin gene in 2 studies (n = 37) [Citation66,Citation67] and lentiviral transduction with the BCH-BB694 vector, which encodes a shRNA inducing erythroid-specific knockdown of BCL11A, in 1 study (n = 3) [Citation68]. Length of follow-up in the HSCT and GT group was 3,881.5 and 58.7 patient-years, respectively. The lack of comparators and the heterogeneity in conditioning regimens and reporting of outcomes precluded meta-analysis. The NOS rating for each study is presented in .
Primary outcomes
Median time to neutrophil (ANC) engraftment post-transplant (defined as >0.5 × 109/L) was reported in 32 studies (HSCT = 29, GT = 3), and ranged from 12.5 to 38 days among HSCT studies and 17 to 26 days among GT studies [Citation13,Citation18,Citation19,Citation21–25,Citation27,Citation28,Citation34,Citation37,Citation39,Citation41,Citation44–46,Citation48,Citation50,Citation52,Citation54,Citation55,Citation57,Citation59–63,Citation65–69]. Median time to platelet engraftment (defined as >50 x109/L) was reported in 24 studies (HSCT = 21, GT = 3), and ranged from 12 to 81 days among HSCT studies, and 22–91 days among GT studies [Citation19,Citation21,Citation24,Citation25,Citation27,Citation31,Citation35,Citation37,Citation39,Citation44–46,Citation50,Citation55,Citation57,Citation59–63,Citation66–69]. Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Transplant-related mortality (TRM) reported in 44/53 HSCT studies was 67/1,051 (6.4%) over a median of 3 years of observation post-HSCT [Citation13,Citation16,Citation18,Citation19,Citation21–28,Citation31,Citation33–44,Citation46–54,Citation56–63,Citation65,Citation70–72]. There was 1 death (1/40, 2.5%) felt to be unrelated to LentiGlobin or transplant more than 18 months post-transplant in the GT group [Citation67]. Graft failure, defined by the European Society for Blood and Marrow Transplantation as ANC <0.5 × 109/L, hemoglobin <80 g/L and platelets <20 x109/L by day +28, or ANC <0.5 × 109/L after initial engraftment not related to relapse, infection, or drug toxicity [Citation73], was 91/993 (9.2%) among 43 HSCT studies reporting this outcome [Citation13,Citation18–20,Citation22–29,Citation31,Citation34–39,Citation41–44,Citation46–50,Citation52–63,Citation65,Citation69,Citation70,Citation74]. There were no reports of graft failure, defined as failure to achieve ANC ≥0.5 × 109/L by day +42[Citation10] in the GT group. Median NOS scores for studies reporting TRM were 2.5 for selection, 0 for comparability, and 3 for outcome. Median NOS scores for graft failure were 3 for selection, 0 for comparability, and 2.5 for outcome.
In HSCT studies, 2-year OS was 91% (N = 192, 8 studies [Citation27,Citation37,Citation46,Citation56–58,Citation64,Citation65]) and 2-year event-free survival (EFS) was 88% (N = 168, 7 studies [Citation27,Citation37,Citation46,Citation56,Citation57,Citation64,Citation65]) (see Table S1 for studies reporting OS/EFS at other time points). OS and EFS at specified time points were not reported for GT studies. Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome for OS, and 3 for selection, 0 for comparability, and 2.5 for outcome for EFS.
GVHD reporting was restricted to the HSCT group, given that GT employs autologous HSCs. Acute GVHD (aGVHD) occurred in 185/979 patients (18.9%) in 47 HSCT studies, with grade I-II aGVHD in 121 patients (12.4%) [Citation16,Citation18,Citation19,Citation21–26,Citation31,Citation33–39,Citation45,Citation49–51,Citation53,Citation54,Citation57,Citation59–63,Citation65,Citation69,Citation75] and grade III-IV aGVHD in 39 patients (4.0%) [Citation16,Citation18,Citation19,Citation23,Citation26,Citation33–35,Citation38,Citation39,Citation57, Citation61,Citation63,Citation64,Citation69,Citation70,Citation74]. GVHD grade was not specified in the remaining patients. Chronic GVHD (cGVHD) rates were reported for 940 patients from 44 HSCT studies, over a median follow-up period of 0.6–19.2 years. Chronic GVHD occurred in 129 patients (13.7%) and was mild-moderate in 64 patients (6.8%)[Citation13,Citation18,Citation19, Citation21,Citation24,Citation27,Citation28,Citation31,Citation34,Citation38,Citation44,Citation50,Citation54,Citation57,Citation61,Citation63,Citation65,Citation75], extensive or severe in 35 patients (3.7%)[Citation13,Citation18,Citation21–24,Citation28,Citation35,Citation38,Citation44,Citation57,Citation61,Citation63,Citation69], and not specified for the remaining 30 patients (3.2%). Median NOS scores for aGVHD and cGVHD were 2 for selection, 0 for comparability, and 3 for outcome.
Twenty-four HSCT studies assessed donor chimerism at 1 year post-transplant [Citation18,Citation21,Citation23–26,Citation31,Citation32,Citation34,Citation36,Citation38,Citation39,Citation41,Citation47,Citation50–52,Citation55–57,Citation59,Citation60,Citation63,Citation64], but only 15 studies quantified the degree of chimerism (raw values [Citation32,Citation41,Citation47,Citation52,Citation55,Citation60], means [Citation31,Citation34,Citation39,Citation63,Citation64], or ranges [Citation50,Citation51,Citation57,Citation59]) (Table S1). Among studies reporting raw chimerism values at 1 year post-HSCT, donor chimerism ranged from 58% to 100% in MRD/MUD transplant recipients who successfully engrafted (n = 41)[Citation32,Citation41,Citation47,Citation52,Citation55,Citation60] and from <5% to 100% in haploidentical transplant recipients with successful engraftment (n = 7)[Citation41]. Median NOS scores for studies reporting 1-year chimerism were 3 for selection, 0 for comparability, and 3 for outcome.
Secondary outcomes
Secondary malignancies
Post-transplant malignancies developed in 6 patients from 5 studies (4 HSCT, 1 GT): in the HSCT group, 2 adult patients who underwent non-myeloablative haploidentical HSCT with total body irradiation and alemtuzumab (1 also received post-transplant cyclophosphamide) developed high-grade myelodysplastic syndrome (MDS) at 2 and 5 years post-transplant [Citation31]. One additional HSCT patient developed MDS, which transformed to acute myeloid leukemia (AML) following a MRD transplant conditioned with busulfan and cyclophosphamide, with possible anti-thymocyte globulin or total lymphoid irradiation (exact conditioning regimen was not reported) [Citation61]. Finally, 2 patients developed post-transplant lymphoproliferative disease (PTLD) in the context of Epstein–Barr virus reactivation [Citation22,Citation64]. Of note, Fitzhugh et al. [Citation31] also reported 1 additional case of PTLD but did not specify whether this was a SCD or thalassemia patient. In the GT studies, 1 patient from the Hgb-206 study developed MDS 36 months post-infusion, which ultimately progressed to AML [Citation76]. He was treated with hydroxyurea for 8 years prior to study enrollment and received LentiGlobin BB305 after busulfan myeloablative conditioning. The MDS/AML was reported as not related to insertional mutagenesis. Duration of pre-transplant exposure to hydroxyurea in the patients who developed malignancy post-transplant was not reported, with the exception of Hsieh et al. [Citation76] The median NOS scores were 3 for selection, 0 for comparability, and 2 for outcome.
Acute chest syndrome & vaso-occlusive episodes
Eighteen studies (16 HSCT [Citation23–25,Citation28,Citation35,Citation36,Citation38,Citation41,Citation46,Citation47,Citation49,Citation54–56,Citation59,Citation69], 2 GT [Citation66,Citation67], n = 345) described the occurrence of ACS and/or VOE post-transplant. While ACS and VOE frequency pre- and post-transplant was generally not reported (thereby precluding any analysis), the vast majority of HSCT studies reported an absence of ACS or VOE in engrafted patients, even with mixed chimerism. One exception is Saraf et al. [Citation56], where 1/12 successfully engrafted patients had 1 VOE post-transplant (median follow-up 1.8 years), though this patient had 12 VOEs/year prior to transplant. Among the GT studies reporting ACS and VOE post-transplant, the Hgb-206 study [Citation67,Citation77] noted a reduction in annualized combined rates of ACS and VOE in 14 Group C patients with prior history of ACS/VOE, from a median of 4.0 (range 2.0-14.0), to 1 VOE and no ACS post-transplant. This patient cohort had a median modified Hgb (HbAT87Q) of 52 g/L (range, 27–94 g/L). Meanwhile, the Hgb-205 study [Citation66,Citation78] reported 2 episodes of ACS and 1 VOE post-GT in a patient with only 7.9% HbAT87Q, as well as one VOE 30 months post-GT in a patient who achieved 48% HbAT87Q. Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Health-related quality of life
Seven studies (6 HSCT [Citation16,Citation17,Citation36,Citation56,Citation57,Citation63,Citation79], 1 GT [Citation67,Citation80]) reported HRQOL outcomes. The HRQOL instruments were heterogeneous and included PedsQL, EQ-5D, CHQ, SF36v1, and PROMIS-57. The time points at which HRQOL was assessed after transplant were also heterogeneous, ranging from single or repeated measures up to 1-year post-transplant [Citation36,Citation56,Citation57,Citation63,Citation67,Citation79], to a single measurement post-transplant with no baseline comparison [Citation17]. Furthermore, studies varied with respect to their use of controls, either with patients on medical therapy, unaffected siblings, or patients acting as their own internal control [Citation16,Citation17,Citation63]. The median NOS score was 3 for selection, 0 for comparability, and 3 for outcome.
While the HRQOL outcomes were too heterogeneous to compare directly, there appears to be a trend toward improvement following HSCT and GT in certain studies. For example, Bhatia et al. [Citation79] reported a mean self-reported PedsQL score change of +16.58 (SE = 5.06) from pre-transplant baseline to one year post-transplant (p = 0.003), and Thompson et al. [Citation67] reported improvements across all domains of PROMIS-57 by 6 months post-GT. However, Arnold et al. [Citation17] demonstrated no statistically significant difference in PedsQL and EQ-5D scores across post-HSCT patients, unaffected siblings, and SCD patients on medical therapy. Similarly, Bernaudin et al. [Citation16] did not note any significant differences between standard of care and HSCT at 1- and 3-years post-transplant in the propensity-matched samples.
Cardiopulmonary outcomes
Eight HSCT (0 GT) studies reported post-transplant cardiac outcomes [Citation18,Citation32,Citation39,Citation48,Citation50,Citation56,Citation63,Citation71,Citation81]. Tricuspid regurgitant jet velocity (TRJV), B-type natriuretic peptide (BNP), and left ventricular (LV) mass index were quantified both pre- and post-transplant in three [Citation48,Citation63,Citation81], two [Citation56,Citation81], and two [Citation71,Citation81] studies, respectively (). However, reporting of other cardiac outcomes – including LV shortening fraction [Citation18], LV strain [Citation81], cardiac output [Citation81], LV ejection fraction [Citation81], and LV end-diastolic volume index [Citation81] – was inconsistent across studies. An overall decrease in TRJV, BNP, and LV mass index was observed post-transplant (). Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Table 3. Summary of cardiac outcomes pre- and post-HSCT reported by more than 1 study.
Ten HSCT (0 GT) studies commented on change in lung function [Citation13,Citation18,Citation24,Citation25,Citation28,Citation39,Citation48,Citation53,Citation56,Citation59,Citation63]. Only 3 studies reported quantitative pulmonary function test (PFT) and 2 reported 6-minute walk test (6MWT) results. While there was no significant difference in PFT parameters, there was an improvement in 6MWT distance for one of the studies (). Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Table 4. Summary of pulmonary outcomes pre- and post-HSCT reported by more than 1 study.
Neurological complications
Six HSCT (0 GT) studies commented on transcranial Doppler (TCD) changes following transplant [Citation16,Citation18,Citation19,Citation25,Citation38,Citation53]. Only 3 (n = 103) reported quantitative TCD velocity data [Citation16,Citation18,Citation38]. There was considerable heterogeneity in the timing and the way TCD data were reported. Across 3 studies, time-averaged mean of maximum velocities (TAMV) was reduced from 138–170 to 81–129 cm/s 1-year post-transplant. Only Bernaudin et al. [Citation16] compared 1-year post-transplant TAMV to standard of care (transfusion) and noted a difference of −39.9 cm/s (p < 0.001). Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Seventeen HSCT studies (n = 433) [Citation13,Citation16,Citation18, Citation19,Citation22,Citation24,Citation25,Citation28,Citation34,Citation38–40,Citation44,Citation47,Citation48,Citation53,Citation59,Citation61,Citation79] reported on the occurrence of CVA post-transplant, defined as stroke, TIA, or cerebral hemorrhage. Among these studies, 124 patients were reported as having stroke or TIA as an indication for transplant. Post-transplant, there were 15 CVAs reported after 2,431.5 patient-years of follow-up, and 2 CVAs reported over an unspecified period of follow-up [Citation44,Citation47]. Two of 17 post-transplant CVAs were ischemic strokes occurring in engrafted patients [Citation44,Citation79] and 10 patients across 8 studies [Citation13,Citation18,Citation19,Citation22,Citation24,Citation28,Citation34,Citation40,Citation79] were noted to have intracranial hemorrhages (ICH) post-transplant. Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Avascular necrosis and nephropathy
Four studies [Citation18,Citation38,Citation61,Citation82] representing 162 patients (3 GT, 159 HSCT) commented on AVN post-transplant. One study [Citation38] reported rapid improvement in AVN findings in 1 patient post-HSCT on MRI, while another [Citation18] reported new AVN in 2 patients at 2 years post-HSCT. A total of 3 studies [Citation18,Citation61,Citation82] (2 HSCT, 1 GT) noted stabilization or no improvement of existing pre-transplant AVN lesions among 7 patients; however, imaging modalities were variable, employing MRI or radiographs. Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Two HSCT studies compared changes in renal function [Citation18,Citation81]. Dallas et al. [Citation18] noted a decrease in mean creatinine clearance from 158±55.7 to 103.5±24 mL/min/1.73 m2 post-MRD-transplant (p = 0.004). A decrease was also noted in the haploidentical group, from 98±33 to 91±47 mL/min/1.73 m2 (no p-value given). Sachdev et al. [Citation81] reported an increase in serum creatinine from 1.0±1.5 mg/dL to 1.4±2.8 mg/dL (p≤0.005) in successful transplants from pre-transplant to 1-year post-transplant. Median NOS scores were 3 for selection, 0 for comparability,and 3 for outcome.
Opioid use
Four HSCT studies [Citation36,Citation41,Citation48,Citation65] (n = 104) commented on changes in opioid requirements post-transplant. Three studies described either discontinuation [Citation41] or a reduction [Citation36,Citation65] in opioid use in some or all patients. Hsieh et al. [Citation48] was the only study to quantify changes in narcotic use: 11 patients on chronic narcotics went from 639 mg IV morphine equivalents/week (95% CI, 220-1, 058) during the week of their transplant to 140 mg (95% CI, 56-225) at six months post-transplant, with 6/11 of these patients being weaned from narcotics altogether. Median NOS scores were 3 for selection, 0 for comparability, and 2.5 for outcome.
Hospitalizations
Six studies [Citation36,Citation46,Citation48,Citation49,Citation59,Citation82] (5 HSCT, 1 GT; n = 89) reported on hospitalization frequency or length of stay pre- and/or post-transplant. Two studies documented hospitalization rates both pre- and post-transplant: Hsieh et al. [Citation48] reported a mean hospitalization rate of 3.23/patient/year pre-transplant (95% CI, 1.83–4.63), which decreased significantly post-transplant, and Ozdogu et al. [Citation36] reported that 18/20 patients had ≥1 hospitalization/year pre-transplant, and only 1/12 patients had ≥1 hospitalization/year post-transplant. Two studies reported post-transplant hospitalization without reference to pre-transplant hospitalization outcomes [Citation46,Citation49]. Median NOS scores were 3 for selection, 0 for comparability, and 3 for outcome.
Discussion
HSCT and GT are both potentially curative options for severe SCD, but each strategy carries unique risks. As there are no clinical trials comparing HSCT to GT, clinicians have little guidance on which strategy to recommend to their patients. Our systematic review sought to compare available data from both strategies across a wide breadth of endpoints, including transplant-related, SCD-specific, and patient-important outcomes.
As expected, the majority of studies were HSCT and duration of follow-up was longer with HSCT, given the recent introduction of GT. Although we were not able to directly compare most outcomes due to a lack of comparator, control group, and heterogeneity in outcome measures, we can draw broad conclusions from our observations. We noted low rates of mortality in both HSCT and GT, with 2-year OS and EFS for the HSCT group at 91% and 88%, respectively, and 2.5% reported mortality in the GT group (limited follow-up duration precluded formal OS and EFS reporting). While we were not able to directly compare graft failure between HSCT and GT due to fundamental differences in the stem cell source (allogeneic versus autologous), their rates appeared to be low (9.2% in the HSCT group, and 0% in the GT group, with 1 GT patient failing to achieve adequate anti-sickling hemoglobin levels). Comparison between HSCT and GT in terms of ‘success’ will remain difficult so long as different metrics are used to determine ‘success’ (for example, donor chimerism in HSCT and vector copy number for GT, and variable reporting of SCD-specific and patient-important outcomes).
Although some SCD-related outcomes demonstrated improvement, such as reduced or absent ACS/VOE post-transplant in both HSCT and GT groups, the lack of a suitable comparator in most studies obfuscated the true effect of HSCT and GT on the outcome. For example, while 2 studies noted improvement in HRQOL scores post-transplant [Citation67,Citation79], 2 others failed to demonstrate a significant difference when compared to standard of care [Citation16,Citation17]. On the other hand, when Bernaudin et al. [Citation16] compared HSCT to transfusion support, they showed a further reduction in TCD by 39.9 cm/s. Likewise, it is difficult to ascertain the impact of transplant on ICH without a suitable control group, since both the transplant and SCD itself are potential contributors. This highlights the need for a comparator group to minimize bias.
One major unanswered question in HSCT and GT is whether these therapies may arrest or reverse end-organ damage sustained pre-transplant, or whether transplant-related toxicity may negate any gains in end-organ function. A minority of HSCT studies assessed change in end-organ function, and even fewer studies quantified these changes. Pulmonary hypertension was improved based on 3 studies that quantified TRJV. PFT results were unchanged in the 3 HSCT studies that reported PFT values, which may be due to a number of possibilities: lung damage that preceded the transplant is irreversible, but no further damage is incurred in the absence of SCD post-HSCT, improvements in lung function were subsequently offset by GVHD, or post-transplant PFT assessment timing was not long enough to detect a clinically meaningful difference. Finally, only 2 studies quantified renal function, which decreased post-transplant. This could be related to conditioning regimen nephrotoxicity, or the normalization of the glomerular filtration rate in response to the amelioration of hyperfiltration typically seen in SCD patients. None of the studies commented on the occurrence of or progression of retinopathy or skin ulcers post-transplant. Again, the lack of comparator limits the interpretation of these findings.
The potential risk of complications from HSCT and GT must be weighed against potential benefits. While the risk of secondary malignancies in the HSCT group is well-established, with 5 reported over more than 3,800 patient-years of follow-up, the relatively early onset of 1 reported case in the GT group over nearly 60 patient-years of follow-up, with an additional case of AML reported since our literature search [Citation83], warrants further examination. While these cases of MDS/AML in the GT group are unlikely to be related to insertional mutagenesis [Citation76,Citation83], the occurrence of secondary malignancies in both treatment groups raises concerns about the consequences of conditioning regimens and highlights the importance of long-term follow-up, as outlined by regulatory agencies such as the FDA [Citation84].
The overall quality of evidence of our included studies was low, with no randomized controlled trials identified from our literature search. There was significant heterogeneity regarding study characteristics and outcomes reporting, both within and between treatment groups. The HSCT studies were highly varied with respect to HSC donor source, participant ages, choice of conditioning regimen, techniques for ascertaining donor chimerism, and timing of chimerism assessments. Reporting of SCD-related outcomes was also highly inconsistent, with studies using different measures for the same outcome. For example, although there appeared to be a trend toward improved HRQOL scores post-transplant in 6 HSCT and 1 GT studies, an observation that was supported by a recent systematic review by Badawy et al. [Citation85], meta-analysis was not possible due to heterogeneity between HRQOL instruments and variable timing of post-transplant assessments. This is in contrast to thalassemia HSCT studies where common standard measures were used, and Badawy et al. [Citation85] were able to quantitatively summarize the findings. Fortunately, many SCD-specific outcomes were specified a priori in GT protocols (e.g. the Hgb-205 study [Citation78], and long term follow-up study NCT02633943 [Citation86]), thus permitting comparison with HSCT in the near future.
Our systematic review has several limitations. While we acknowledge that younger age, certain HSCT donor sources/conditioning regimens, and more recent year of HSCT are associated with more favorable outcomes [Citation87,Citation88], the intent of this systematic review was to compare two broad methodologies of potentially curative options. The availability of detailed transplant registries provides a more efficient approach in answering questions related to age, HSCT donor sources, conditioning regimens, and year of transplant. As such, we did not perform extensive subgroup analyzes comparing these parameters. One group has attempted such a comparison, but results are not yet available owing to only 1 ongoing trial that met the inclusion/exclusion criteria of the systematic review [Citation89]. Another limitation of our study is the small sample size and short duration of follow-up for the GT group, which precluded further subgroup analyses of the different GT techniques (e.g. CRISPR/Cas9 or lentiglobin vector), which in and of themselves may produce different outcomes. However, this systematic review will serve as a foundation for future iterations when more data become available.
Although HSCT and GT methodologies share comparable short-term mortality and improvement in certain SCD-related outcomes, long-term complications, such as secondary malignancies, can potentially be devastating. For SCD patients, caregivers, and clinicians to make informed decisions in choosing one therapy over another, accurate estimates of the comparative risks and benefits are needed. This in turn predicates on being able to compare the same outcome measures across different studies and techniques. We thus advocate for standardized reporting to better compare outcomes within and between treatment groups, to allow for more informed clinical decision-making when referring patients with SCD for potentially curative therapy.
Acknowledgements
The authors are grateful to Dr. Gordon Guyatt (McMaster University, Hamilton, Canada) for his guidance and support. L.E.R. and K.H.M.K. designed the study; L.E.R. and C.J.P. performed screening for study inclusion and exclusion; L.E.R. and K.H.M.K. performed risk of bias grading; L.E.R. extracted and analyzed the data; L.E.R., K.H.M.K., A.V., and R.K. interpreted the data; L.E.R. drafted the manuscript; L.E.R., K.H.M.K., A.V., R.K., and C.J.P. critically revised the paper. All authors approved of the final version of the manuscript.
Disclosure statement
C.J.P.: Advisory Board Honoraria: Alexion, Apellis, BioCryst, Octapharma, Sanofi. Speaking Honoraria: Alexion, Octapharma. K.H.M.K.: Consultancy: Agios, Alexion, Apellis, Aruvant, Bluebird Bio, Celgene/BMS, Forma, Pfizer, Novartis; Research funding: Pfizer, NHLBI/NIH; Membership on an entity's Board of Directors or advisory committees: Bioverativ/Sanofi, Research collaboration: Phoenicia Biosciences, Abfero.
Additional information
Funding
References
- Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89(5):530–535.
- Gardner K, Douiri A, Drasar E, et al. Survival in adults with sickle cell disease in a high-income setting. Blood. 2016;128(10):1436–1438.
- Bauer DE, Brendel C, Fitzhugh CD. Curative approaches for sickle cell disease: a review of allogeneic and autologous strategies. Blood Cells Mol Dis. 2017;67:155–168.
- Guilcher GMT, Truong TH, Saraf SL, et al. Curative therapies: allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and non-myeloablative conditioning in sickle cell disease. Semin Hematol. 2018;55(2):87–93.
- Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016;127(7):839–848.
- Esrick EB, Brendel C, Manis JP, et al. Flipping the switch: initial results of genetic targeting of the fetal to adult globin switch in sickle cell patients [abstract]. Blood. 2018;132(suppl 1):1023.
- Hsieh MM, Fitzhugh C, Tisdale J. Allogeneic hematopoietic stem cell transplantation for sickle cell disease: the time is now. Blood. 2011;118(5):1197–1207.
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62(10):1006–1012.
- Carreras E, Dufour C, Mohty M, Kroger N, editors. The EBMT handbook: hematopoietic stem cell transplantation and cellular therapies. Cham: Springer Open; 2019.
- Kanter J, Walters MC, Krishnamurti L, et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med. 2022;386(7):617–628.
- Stenger E, Phelan R, Shaw BE, et al. Excellent overall survival and low incidence of late effects in patients undergoing allogeneic hematopoietic cell transplant for sickle cell disease: a report from the Center for International Blood and Marrow Transplant Research (CIBMTR). Blood. 2019;134(suppl 1):697.
- Cochrane handbook for systematic reviews of interventions, Version 5.1.0. 16.1.3.2 Imputing standard deviations for changes from baseline [Internet]. Publication date: 2011 [cited 2022 Apr 20]. 2011 [Available from: https://handbook-5-1.cochrane.org/chapter_16/16_1_3_2_imputing_standard_deviations_for_changes_from_baseline.htm].
- Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263–272.
- Cochrane handbook for systematic reviews of interventions, Version 5.1.0. 7.7.3.2 Obtaining standard deviations from standard errors and confidence intervals for group means [Internet]. Publication date: 2011 [cited 2022 May 12]. 2011 [Available from: http://handbook-5-1.cochrane.org/chapter_7/7_7_3_2_obtaining_standard_deviations_from_standard_errors_and.htm].
- Wells GA, Shea B, O’Connell D, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses [Internet]. Ottawa (CAN): Ottawa Hospital Research Institute; Publication date unknown [cited 2021 Mar 17]. Available from: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp.
- Bernaudin F, Verlhac S, De Latour RP, et al. Association of matched sibling donor hematopoietic stem cell transplantation with transcranial Doppler velocities in children with sickle cell anemia. JAMA. 2019;321(3):266–276.
- Arnold SD, Jin Z, Sands S, et al. Allogeneic hematopoietic cell transplantation for children with sickle cell disease is beneficial and cost-effective: a single-center analysis. Biol Blood Marrow Transplant. 2015;21(7):1258–1265.
- Dallas MH, Triplett B, Shook DR, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2013;19(5):820–830.
- Dedeken L, Le PQ, Azzi N, et al. Haematopoietic stem cell transplantation for severe sickle cell disease in childhood: a single centre experience of 50 patients. Br J Haematol. 2014;165(3):402–408.
- Bazuaye NG, Enomamien B. Hematopoietic stem cell transplantation (HSCT) for hemoglobinopathy (sickle cell disease) in Nigeria-2013 update by Nigerian group for blood and marrow transplant (NGBMT) [abstract]. Bone Marrow Transplant. 2014;49:S320–S3S1.
- Horan JT, Haight A, Dioguardi JL, et al. Using fludarabine to reduce exposure to alkylating agents in children with sickle cell disease receiving busulfan, cyclophosphamide, and antithymocyte globulin transplant conditioning: results of a dose de-escalation trial. Biol Blood Marrow Transplant. 2015;21(5):900–905.
- Kamani NR, Walters MC, Carter S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the Phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol Blood Marrow Transplant. 2012;18(8):1265–1272.
- Lucarelli G, Isgro A, Sodani P, et al. Hematopoietic SCT for the Black African and non-Black African variants of sickle cell anemia. Bone Marrow Transplant. 2014;49(11):1376–1381.
- Majumdar S, Robertson Z, Robinson A, et al. Outcome of hematopoietic cell transplantation in children with sickle cell disease, a single center’s experience. Bone Marrow Transplant. 2010;45(5):895–900.
- Marzollo A, Calore E, Tumino M, et al. Treosulfan-based conditioning regimen in sibling and alternative donor hematopoietic stem cell transplantation for children with sickle cell disease. Mediterr J Hematol Infect Dis. 2017;9(1):e2017014.
- McPherson ME, Hutcherson D, Olson E, et al. Safety and efficacy of targeted busulfan therapy in children undergoing myeloablative matched sibling donor BMT for sickle cell disease. Bone Marrow Transplant. 2011;46(1):27–33.
- Radhakrishnan K, Bhatia M, Geyer MB, et al. Busulfan, fludarabine, and alemtuzumab conditioning and unrelated cord blood transplantation in children with sickle cell disease. Biol Blood Marrow Transplant. 2013;19(4):676–677.
- Walters MC, Storb R, Patience M, et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Blood. 2000;95(6):1918–1924.
- Woodard P, Helton KJ, Khan RB, et al. Brain parenchymal damage after haematopoietic stem cell transplantation for severe sickle cell disease. Br J Haematol. 2005;129(4):550–552.
- Zhao J, Beebe K, Magee K, et al. Adolescent male fertility following reduced-intensity conditioning regimen for hematopoietic stem cell transplantation in non-malignant disorders. Pediatr Transplant. 2019;23(6):e13496.
- Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017;1(11):652–661.
- Horwitz ME, Spasojevic I, Morris A, et al. Fludarabine-based nonmyeloablative stem cell transplantation for sickle cell disease with and without renal failure: clinical outcome and pharmacokinetics. Biol Blood Marrow Transplant. 2007;13(12):1422–1426.
- Oevermann L, Zimmermann C, Voigt S, et al. Transmission of chromosomally integrated human herpes virus-6A via haploidentical stem cell transplantation poses a risk for virus reactivation and associated complications. Bone Marrow Transplant. 2020;55(1):260–264.
- Saraf S, Oh AL, Patel P, et al. Haploidentical peripheral blood stem cell transplantation demonstrates stable engraftment in adults with sickle cell disease. Biol Blood Marrow Transplant. 2018;24(8):1759–1765.
- van Besien K, Bartholomew A, Stock W, et al. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2000;26(4):445–449.
- Ozdogu H, Boga C, Yeral M, et al. Allogenic peripheral stem cell transplantation from HLA-matched related donors for adult sickle cell disease: remarkable outcomes from a single-center trial. Bone Marrow Transplant. 2018;53(7):880–890.
- Alzahrani M, Damlaj M, Essa M, et al. Outcome of age-adapted approach to HLA-identical related hematopoietic stem cell transplantation in severe sickle cell disease: Saudi experience [abstract]. Blood. 2018;132(suppl 1):3468.
- Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749–2756.
- Bhatia M, Jin Z, Baker C, et al. Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant. 2014;49(7):913–920.
- Bhunia N, Abu-Arja R, Bajwa RPS, et al. Successful treatment with eculizumab for posterior reversible encephalopathy syndrome due to underlying transplant-associated thrombotic microangiopathy in patients transplanted for sickle cell disease. Pediatr Blood Cancer. 2019;66(10):e27912.
- Bolanos-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285–4291.
- De Santis GC, Costa TCM, Santos FLS, et al. Blood transfusion support for sickle cell patients during haematopoietic stem cell transplantation: a single-institution experience. Br J Haematol. 2020;190(5):e295–e2e7.
- Dhedin N, de la Fuente J, Bernaudin F, et al. Haploidentical bone marrow transplant with post-transplant cytoxan plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international multicenter learning collaborative [abstract]. Blood. 2016;128(22):1233.
- Eckrich MJ, Madden L, Taylor C, et al. Reduced-intensity allogeneic stem cell transplant for sickle cell disease in pediatrics: a single institution experience [abstract]. Biol Blood Marrow Transplant. 2018;24(3):S434–S4S5.
- Frangoul H, Evans M, Isbell J, et al. Haploidentical hematopoietic stem cell transplant for patients with sickle cell disease using thiotepa, fludarabine, thymoglobulin, low dose cyclophosphamide, 200 cGy TBI and post transplant cyclophosphamide. Bone Marrow Transplant. 2018;53(5):647–650.
- Guilcher GMT, Monagel DA, Nettel-Aguirre A, et al. Nonmyeloablative matched sibling donor hematopoietic cell transplantation in children and adolescents with sickle cell disease. Biol Blood Marrow Transplant. 2019;25(6):1179–1186.
- Horan JT, Liesveld JL, Fenton P, et al. Hematopoietic stem cell transplantation for multiply transfused patients with sickle cell disease and thalassemia after low-dose total body irradiation, fludarabine, and rabbit anti-thymocyte globulin. Bone Marrow Transplant. 2005;35(2):171–177.
- Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48–56.
- Iannone R, Casella JF, Fuchs EJ, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519–528.
- Krishnamurti L, Kharbanda S, Biernacki MA, et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2008;14(11):1270–1278.
- Kuentz M, Robin M, Dhedin N, et al. Is there still a place for myeloablative regimen to transplant young adults with sickle cell disease? Blood. 2011;118(16):4491–4492.
- Matthes-Martin S, Lawitschka A, Fritsch G, et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur J Haematol. 2013;90(4):308–312.
- Mynarek M, Bettoni Da Cunha Riehm C, Brinkmann F, et al. Normalized transcranial Doppler velocities, stroke prevention and improved pulmonary function after stem cell transplantation in children with sickle cell anemia. Klin Padiatr. 2013;225(3):127–132.
- Pawlowska AB, Cheng JC, Karras NA, et al. HLA haploidentical stem cell transplant with pretransplant immunosuppression for patients with sickle cell disease. Biol Blood Marrow Transplant. 2018;24(1):185–189.
- Rangarajan HG, Abu-Arja R, Pai V, et al. Outcomes of unrelated donor stem cell transplantion with post-transplant cyclophosphamide for graft-versus-host disease prophylaxis in patients with severe sickle cell disease. Biol Blood Marrow Transplant. 2018;24(2):413–417.
- Saraf SL, Oh AL, Patel PR, et al. Nonmyeloablative stem cell transplantation with alemtuzumab/low-dose irradiation to cure and improve the quality of life of adults with sickle cell disease. Biol Blood Marrow Transplant. 2016;22(3):441–448.
- Shenoy S, Eapen M, Panepinto J, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016;128(21):2561–2567.
- Shyr D, Leung K, Krance R, et al. A single center experience using myeloablative hematopoietic stem cell transplantation for beta-thalassemia and sickle cell disease [abstract]. Pediatr Blood Cancer. 2012;58(7):1022.
- Soni S, Gross TG, Rangarajan H, et al. Outcomes of matched sibling donor hematopoietic stem cell transplantation for severe sickle cell disease with myeloablative conditioning and intermediate-dose of rabbit anti-thymocyte globulin. Pediatr Blood Cancer. 2014;61(9):1685–1689.
- Strocchio L, Zecca M, Comoli P, et al. Treosulfan-based conditioning regimen for allogeneic haematopoietic stem cell transplantation in children with sickle cell disease. Br J Haematol. 2015;169(5):726–736.
- Vermylen C, Cornu G, Ferster A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998;22(1):1–6.
- Wiebking V, Hutker S, Schmid I, et al. Reduced toxicity, myeloablative HLA-haploidentical hematopoietic stem cell transplantation with post-transplantation cyclophosphamide for sickle cell disease. Ann Hematol. 2017;96(8):1373–1377.
- Krishnamurti L, Neuberg D, Sullivan K, et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: results of a prospective multicenter pilot study. Am J Haematol. 2019;94(4):446–454.
- Chaudhury S, Volonte B, Rangarajan HG, et al. Abatacept for GVHD prophylaxis after hematopoietic cell transplantation (HCT) for pediatric sickle cell disease (SCD): a sickle cell Transplant Advocacy and Research Alliance (STAR) trial [abstract]. Biol Blood Marrow Transplant. 2020;26(S20).
- Foell J, Kleinschmidt K, Jakob M, et al. Alternative donor: alphas/CD19 T-cell-depleted haploidentical hematopoietic stem cell transplantation for sickle cell disease. Hematol Oncol and Stem Cell Ther. 2020;13(2):98–105.
- Magrin E, Semeraro M, Magnani A, et al. Results from the completed HGB-205 trial of lentiglobin for beta-thalassemia and lentiglobin for sickle cell disease gene therapy [abstract]. Blood. 2019;134:3358.
- Thompson AA, Walters MC, Mapara MY, et al. Resolution of serious vaso-occlusive pain crises and reduction in patient-reported pain intensity: results from the ongoing phase 1/2 HGB-206 Group C Study of LentiGlobin for sickle cell disease (bb1111) gene therapy [abstract]. Blood. 2020;136:16–17.
- Esrick EB, Achebe M, Armant M, et al. Validation of BCL11A as therapeutic target in sickle cell disease: results from the adult cohort of a pilot/feasibility gene therapy trial inducing sustained expression of fetal hemoglobin using post-transcriptional gene silencing [abstract]. Blood. 2019;134(Suppl 2):LBA5.
- Adamkiewicz TV, Mehta PS, Boyer MW, et al. Transplantation of unrelated placental blood cells in children with high-risk sickle cell disease. Bone Marrow Transplant. 2004;34(5):405–411.
- Abraham A, Stenger E, Travers CD, et al. Similar event-free survival but higher incidence of mixed T cell chimerism after hematopoietic cell transplantation for sickle cell disease using rabbit versus horse ATG: A study from the Sickle Transplant Alliance for Research (STAR) [abstract]. Blood. 2017;130(Suppl 1):3535.
- Covi S, Ravindranath Y, Farooqi A, et al. Changes in bi-ventricular function after hematopoietic stem cell transplant as assessed by speckle tracking echocardiography. Pediatr Cardiol. 2018;39(2):365–374.
- Lucarelli G, Gaziev J, Isgro A, et al. Allogeneic cellular gene therapy in hemoglobinopathies-evaluation of hematopoietic SCT in sickle cell anemia. Bone Marrow Transplant. 2012;47(2):227–230.
- Valcarcel D, Sureda A. Chapter 41: graft failure. The EBMT handbook: hematopoietic stem cell transplantation and cellular therapies [internet]. 7th Ed. Carreras E, Dufour C, Mohty M, editors. Cham (CH): Springer; 2019.
- Al-Musalhi M, Al-Zadjali S, Al-Huneini M, et al. Stable mixed chimerism after hematopoietic stem cell transplantation in patients with sickle cell disease [abstract]. Bone Marrow Transplant. 2017;52(Suppl 1):96.
- Biernacki MA, Okam M, Shenoy S, et al. Long-term follow-up of adults with severe sickle cell disease after hematopoietic stem cell transplantation using reduced intensity conditioning [abstract]. Blood. 2010;116(21):261.
- Hsieh MM, Bonner M, Pierciey FJ, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4(9):2058–2063.
- ClinicalTrials.gov. A study evaluating the safety and efficacy of bb1111 in severe sickle cell disease [NCT02140554] [updated December 29, 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT02140554].
- ClinicalTrials.gov. A study evaluating the safety and efficacy of LentiGlobin BB305 Drug Product in β-thalassemia major (also referred to as transfusion-dependent β-thalassemia [TDT]) and sickle cell disease [NCT02151526] [updated April 26, 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT02151526].
- Bhatia M, Kolva E, Cimini L, et al. Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2015;21(4):666–672.
- Kanter J, Tisdale J, Mapara M, et al. Improvements in health-related quality of life for patients treated with LentiGlobin for sickle cell disease (bb1111) gene therapy [abstract]. Blood. 2020;136:365.
- Sachdev V, Hsieh M, Jeffries N, et al. Reversal of a rheologic cardiomyopathy following hematopoietic stem cell transplantation for sickle cell disease. Blood Adv. 2019;3(19):2816–2824.
- Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848–855.
- Goyal S, Tisdale J, Schmidt M, et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med. 2022;386:138–147.
- Food and Drug Administration Center for Biologics Evaluation and Research. Long term follow-up after administration of human gene therapy products: guidance for industry. 2020 [updated January 2020. Available from: https://www.fda.gov/media/113768/download].
- Badawy SM, Beg U, Liem RI, et al. A systematic review of quality of life in sickle cell disease and thalassemia after stem cell transplant or gene therapy. Blood Adv. 2021;5(2):570–583.
- ClinicalTrials.gov. Longterm follow-up of subjects with hemoglobinopathies treated with ex vivo gene therapy [NCT02633943] [updated April 26, 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT02633943].
- Eapen M, Brazauskas R, Walters MC, et al. Impact of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with Sickle Cell Disease: a retrospective, cohort study. Lancet Haematol. 2019;6(11):e585–ee96.
- Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129(11):1548–1556.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev. 2020;7(7):CD007001.
- Dennison D, Al Farsi K, Al Huneini M, et al. Hematopoietic stem cell transplantation for sickle cell anemia with busulfan-based reduced intensity conditioning: cure and fertility [abstract]. Biol Blood Marrow Transplant. 2013;19(2):S285–S2S6.
- Williams KM, Dietzen D, Hassoun AA, et al. Autoimmune thyroid disease following alemtuzumab therapy and hematopoietic cell transplantation in pediatric patients with sickle cell disease. Pediatr Blood Cancer. 2014;61(12):2307–2309.
- Bhunia N, Abu-Arja R, Bajwa RPS, et al. Eculizumab to treat posterior reversible encephalopathy syndrome due to underlying transplant-associated thrombotic microangiopathy in patients receiving allogeneic hematopoietic cell transplant for sickle cell disease [abstract]. Biol Blood Marrow Transplant. 2019;25(S140).
- Chaudhury S, Laskowski J, Rangarajan HG, et al. Abatacept for GVHD prophylaxis after hematopoietic stem cell transplantation (HCT) for pediatric sickle cell disease (SCD): A sickle transplant alliance for research (STAR) trial [abstract]. Biol Blood Marrow Transplant. 2018;24(3):S91.
- Covi S, Ravindranath Y, Farooqi A, et al. Effect of hematopoietic stem cell transplantation on bi-ventricular function as assessed by speckle tracking echocardiography [abstract]. J Am Soc Echocardiogr. 2016;29(6):B92.
- Dedeken L, Le PQ, Azzi N, et al. Hematopoietic stem cell transplantation for sickle cell disease in childhood: A single center experience with 45 patients [abstract]. Blood. 2011;118(21):3103.
- Brachet C, Azzi N, Demulder A, et al. Hydroxyurea treatment for sickle cell disease: impact on haematopoietic stem cell transplantation’s outcome. Bone Marrow Transplant. 2004;33(8):799–803.
- Brachet C, Heinrichs C, Tenoutasse S, et al. Children with sickle cell disease: growth and gonadal function after hematopoietic stem cell transplantation. J Pediatr Hematol Oncol. 2007;29(7):445–450.
- Foell J, Schulte J, Troeger A, et al. Stem cell transplantation in advanced stage sickle cell disease with haploidentical T-cell depleted PBSC yields comparable outcomes to matched sibling donor bone marrow: results of a pilot study [abstract]. Blood. 2018;132:3455.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361(24):2309–2317.
- Isgro A, Diomedi M, Sodani P, et al. Hematopoietic stem cell transplantation for the black African variants of sickle cell anemia: TCD and brain MRI before transplant [abstract]. Cerebrovasc Dis. 2014;37:42–43.
- Isgro A, Gaziev J, Sodani P, et al. Hematopoietic stem cell transplantation in Nigerian children with sickle cell anemia [abstract]. Blood. 2015;126(23):4598.
- Isgro A, Gaziev J, Sodani P, et al. Hematopoietic stem cell transplantation in Nigerian children with sickle cell anemia [abstract]. Bone Marrow Transplant. 2016;51:S254–S2S5.
- Isgro A, Paciaroni K, Gaziev J, et al. Hematopoietic stem cell transplantation in sickle cell anemia children patients [abstract]. Bone Marrow Transplant. 2015;50:S46.
- Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Update on the first patients with severe hemoglobinopathies treated with lentiglobin gene therapy [abstract]. Haematologica. 2017;102:248.
- Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Update from the Hgb-205 phase 1/2 clinical study of lentiglobin gene therapy: sustained clinical benefit in severe hemoglobinopathies [abstract]. Blood. 2016;128(22):2311.
- Cavazzana M, Hacein-Bey-Abina S, Payen E, et al. Longer term follow-up on the first patients with severe hemoglobinopathies treated with lentiglobin gene therapy [abstract]. Blood. 2017;130(Suppl 1):4609.
- Cavazzana M, Ribeil JA, Payen E, et al. Outcomes of gene therapy for severe sickle disease and beta-thalassemia major via transplantation of autologous hematopoietic stem cells transduced ex vivo with a lentiviral beta AT87Q-globin vector [abstract]. Blood. 2015;126(23):202.
- Cavazzana M, Ribeil JA, Payen E, et al. Outcomes of gene therapy for b-thalassemia major and severe sickle disease via transplantation of autologous hematopoietic stem cells transduced ex vivo with a lentiviral beta globin vector [abstract]. Haematologica. 2015;100:171.
- Cavazzana M, Ribeil JA, Payen E, et al. Clinical outcomes of gene therapy with BB305 lentiviral vector for sickle cell disease and beta-thalassemia [abstract]. Mol Ther. 2016;24:S111–S1S2.
- Hebert N, Magrin E, Miccio A, et al. Analysis of RBC properties in patients with SCD treated with lentiglobin gene therapy [abstract]. Blood. 2018;132(Suppl 1):2195.
- Soni S, Tisdale JF, Hsieh M, et al. Outcomes of gene therapy for severe sickle cell disease (SCD) via transplantation of autologous hematopoietic stem cells transduced Ex vivo with a lentiviral beta AT87Q-globin vector. Biol Blood Marrow Transplant. 2016;22(3):S63.
- King AA, McKinstry RC, Wu J, et al. Functional and radiologic assessment of the brain after reduced-intensity unrelated donor transplantation for severe sickle cell disease: blood and Marrow Transplant Clinical Trials Network Study 0601. Biol Blood Marrow Transplant. 2019;25(5):e174–e1e8.
- Thompson AA, Tisdale JF, Kanter J, et al. Lentiglobin gene therapy in patients with sickle cell disease: updated interim results from HGB-206 [abstract]. Br J Haematol. 2019;185:22–23.
- Mapara MY, Tisdale JF, Kanter J, et al. Lentiglobin gene therapy in patients with sickle cell disease: updated interim results from Hgb-206 [abstract]. Biol Blood Marrow Transplant. 2019;25:S64–SS5.
- Kanter J, Thompson A, Mapara M, et al. Recent progress in gene therapy for severe sickle cell disease: updated interim results from a phase 1 clinical study of lentiglobin gene therapy. HemaSphere. 2018;2(Suppl 2):368.
- Kanter J, Walters MC, Hsieh M, et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease [abstract]. Blood. 2017;130(Suppl 1):527.
- Kanter J, Walters MC, Hsieh MM, et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease [abstract]. Blood. 2016;128(22):1176.
- Kanter J, Walters MC, Hsieh M, et al. Initial results from study HGB-206: A phase 1 study evaluating gene therapy by transplantation of autologous CD34+ stem cells transduced ex vivo with the lentiglobin BB305 lentiviral vector in subjects with severe sickle cell disease. Blood. 2015;126(23):3233.
- Tisdale JF, Kanter J, Hsieh M, et al. Single-agent plerixafor mobilization to collect autologous stem cells for use in gene therapy for severe sickle cell disease [abstract]. Biol Blood Marrow Transplant. 2018;24(3):S174.
- Tisdale JF, Pierciey FJ, Kamble R, et al. Successful plerixafor-mediated mobilization, apheresis, and lentiviral vector transduction of hematopoietic stem cells in patients with severe sickle cell disease [abstract]. Blood. 2017;130:990.
- Bonner M, Kanter J, MacAri E, et al. The relationships between target gene transduction, engraftment of HSCs and RBC physiology in sickle cell disease gene therapy [abstract]. Blood. 2019;134:206.
- Kanter J, Tisdale JF, Mapara MY, et al. Resolution of sickle cell disease manifestations in patients treated with lentiglobin gene therapy: updated results from the phase 1/2 hgb-206 group c study [abstract]. Blood. 2019;134:990.
- Walters MC, Kanter J, Kwiatkowski JL, et al. Lentiglobin for sickle cell disease (SCD) gene therapy (GT): updated results in group C patients from the phase 1/2 Hgb-206 study [abstract]. Biol Blood Marrow Transplant. 2020;26:S1–S2.
- Walters MC, Tisdale JF, Kwiatkowski JL, et al. Exploring the drivers of potential clinical benefit in initial patients treated in the Hgb-206 study of lentiglobin for sickle cell disease (SCD) gene therapy [abstract]. Blood. 2019;134:2061.