
ABSTRACT
Objective:
Proinflammatory cytokines are considered to be one of the key causes of haemophilic cartilage destruction by inducing chondrocyte apoptosis and extracellular matrix degradation. However, few studies have focused on how proinflammatory cytokines regulate the phenotypic changes of chondrocytes, which may be an important factor in haemophilic cartilage degradation pathogenesis. More understanding is needed about the effect of proinflammatory cytokines on phenotypic changes of the chondrocyte. The objective of this study was to examine how IL-6, TNF-α and IL-1β regulate the chondrocyte phenotype, which may be an important factor in haemophilic cartilage degradation pathogenesis.
Methods:
HUM-iCell-s018 chondrocytes were treated with increasing concentrations of TNF-α, IL-6 or IL-1β (0, 1, 5, 10 ng/ml) for 24 h, then FGF23 and SOX9 expression was determined by qRT-PCR and WB, respectively.
Results:
We found that TNF-α, IL-6 and IL-1β induced FGF23 and suppressed SOX9 expression in chondrocytes in a dose-dependent manner. IL-1β had a stronger regulatory effect on FGF23, while TNF-α and IL-6 had stronger regulatory effects on SOX9.
Conclusions:
These findings suggest that IL-6, IL-1β and TNF-α may be involved in haemophilic cartilage destruction pathogenesis by altering the chondrocyte phenotype through modulation of FGF23 and SOX9 gene expression.
1. Introduction
Haemophilia is a rare congenital, recessive X-linked disorder caused by a deficiency or lack of coagulation factors Ⅸ (FⅨ) or Ⅷ (FⅧ) [Citation1]. Hemarthrosis is a condition caused by recurrent joint bleeding due to coagulation factor deficiency, which can induce the destruction of structures within the joint, such as the menisci, bone and cartilage. This condition can ultimately result in haemophilic arthritis (HA) characterized by pain, deformity and dysfunction of the joints at early stage, and arthrofibrosis, the loss of joint function and disability in the advanced stage [Citation2].
Destruction of articular cartilage is one of the main characteristics of HA. Proinflammatory cytokines, especially IL-6, TNF-α and IL-1β, are involved in many of the body's pathological mechanisms and are considered to be one of the key causes of haemophilic cartilage destruction [Citation3]. They induce cartilage degradation indirectly by destroying extracellular matrix (ECM) by affecting the glycosaminoglycan (GAG) metabolism and upregulating the expression of MMP-13 via activation of the NF-κB signalling pathway, ultimately leading to HIF2-α overexpression. They also lead to cartilage degradation directly by inducing chondrocyte apoptosis by forming hydroxyl radicals in the vicinity of chondrocytes [Citation2]. However, few studies have examined the effect of proinflammatory cytokines on the chondrocyte phenotype, which may be an important consideration in the pathogenesis of haemophilic cartilage degradation.
FGF23 is a protein that is a member of FGF19 subfamily which has been widely studied in phosphate and vitamin D metabolism over the last few decades [Citation4–6]. This secretory protein has also been confirmed to play a crucial part in regulating cartilage metabolism. Excess FGF23 has a strong inhibitory effect on chondrocyte proliferation. FGF23 overload has been shown to upregulate MMP13 expression in chondrocytes resulting in cartilage ECM degradation and subsequent cartilage destruction [Citation7,Citation8]. SOX9, a member of the SOX (SRY-related HMG-box) family that is mainly secreted by chondrocytes, could also be a vital regulator of the cartilage metabolic cycle [Citation9,Citation10]. Low levels of SOX9 in cartilage may inhibit chondrogenesis, cartilage regeneration and differentiation [Citation11]. Additionally, down-regulated expression of SOX9 may upregulate the MMP13 expression in chondrocytes [Citation8]. In a previous study, we found higher levels of FGF23 and lower levels of SOX9 in HA cartilage compared with osteoarthritis (OA), and showed that the expression of FGF23 and SOX9 affected iron regulation in chondrocytes [Citation12]. Like iron, proinflammatory cytokines also have a key role in HA cartilage degradation pathogenesis, but their relationship to phenotype alterations in chondrocytes remains unclear.
Approximately 80% of patients with severe hemophilia live in developing countries, where economic and infrastructure related constraints are more likely to prevent access to adequate prophylaxis [Citation13,Citation14]. HA is one of the most common complications in those patients. However, even with standardized treatment, it's difficult to completely avoid the development of late joint disease [Citation15,Citation16]. In order to delay joint cartilage damage and restore normal joint function, it's necessary to explore the mechanisms of HA cartilage destruction and identify possible new therapies.
In this study, we aimed to investigate the effects of proinflammatory cytokines on the chondrocyte phenotype. We explored IL-6, TNF-α and IL-1β regulation of FGF23 and SOX9 expression in chondrocytes in vitro by gene and protein detection. Determining the effect of proinflammatory cytokines on phenotype changes in chondrocytes would facilitate a better understanding of haemophilic cartilage degradation pathogenesis and provide insight into new avenues for treatment and prevention.
2. Materials and methods
2.1. Regents
Recombinant human IL-1β (CG93, 0332225ED02) and IL-6 (C009, 0332363JE30) were purchased from Novoprotein (Suzhou, China). Recombinant human TNF-α (HY-p7058, 80693) was purchased from MCE (Shanghai, China). SOX9 (USA, AF6330, diluted 1:500) antibody and FGF23 (USA, DF3596, diluted 1:500) antibody were purchased from Affinity.
2.2. Cell culture
Primary human normal chondrocyte cells (HUM-iCell-s018) were purchased from iCell Bioscience Inc. Cells were cultured in ICell Primary Chondrocyte Culture System (PriMed-iCell-020) in a humidified, 5% CO2 atmosphere at 37°C. Chondrocytes were treated by gradient concentrations of IL-6, TNF-α or IL-1β (0, 1, 5, 10 ng/ml) for 24 h before cells were seeded in 6-well plates at a density of 5 × 105 cells/well for 16 h. In order to preserve the chondrocyte phenotype, cells were used without subculturing in all assays.
2.3. Quantitative real-time polymerase chain reaction and Western blot
After chondrocytes were treated by gradient concentrations of IL-6, TNF-α or IL-1β, cells were collected for assessment of gene and protein expression of FGF23 and SOX9 by qRT-PCR and WB, respectively. The experimental procedures for qRT-PCR and WB were conducted as described before [Citation12]. The primers used are presented in .
Table 1. Primers designed for quantitative real-time PCR.
2.4. Statistical analysis
All analyses were performed with SPSS software (version 24.0). The Shapiro–Wilk test was applied in order to verify the normality of the distribution. Student's t-test was used for two group comparisons. Results are presented as effect size with 95% confidence interval (CI). P-values < 0.05 were considered to be significant.
3. Results
3.1. IL-6, TNF-α and IL-1β upregulate the FGF23 expression in chondrocytes
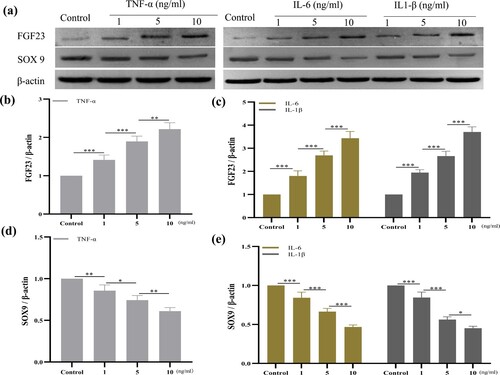
As shown in (a) and (a–c), TNF-α, IL-6 and IL-1β upregulated the expression of FGF23 in chondrocytes in a dose-dependent manner. TNF-α:0 vs 1, p < 0.001 (−1.608, 95%CI [−2.064, −1.152]); 1 vs 5 p < 0.001 (−1.198, 95%CI [−1.654, −0.7419]); 5 vs 10 p < 0.001 (−2.586, 95%CI [−3.042, −2.130]); IL-6: 0 vs 1 p < 0.001 (−1.253, 95%CI [−1.635, −0.8705]); 1 vs 5 p < 0.001 (−1.232, 95%CI [−1.614, −0.8496]); 5 vs 10 p < 0.001 (−2.417, 95%CI [−2.799, −2.035]); IL-1β: 0 vs 1 p < 0.001 (−2.319, 95%CI [−2.890, −1.748]); 1 vs 5 p < 0.001 (−1.184, 95%CI [−1.755, −0.6132]); 5 vs 10 p < 0.001 (−2.238, 95%CI [−2.809, −1.667]). In order to determine which cytokines had the strongest regulatory effect on chondrocytes, we compared the expression of FGF23 at identical concentrations treated with TNF-α, IL-6 or IL-1β, respectively ((b)). The stimulating effect of IL-1β on chondrocytes was stronger than that of TNF-α and IL-6 at 1 and 5 ng/ml, but at 10 ng/ml, it was stronger than IL-6 only; 1 ng/ml:TNF-α vs IL-1β: P = 0.0156 (−0.7113, 95%CI [−1.256, −0.1667]); IL-6 vs IL-1β: P = 0.002 (−1.066, 95%CI [−1.611, −0.5217]); 5 ng/ml: TNF-α vs IL-1β: P = 0.0175 (−0.6977, 95%CI [−1.246, −0.1499]); IL-6 vs IL-1β: P = 0.0027 (−1.019, 95%CI [−1.566, −0.4708]); 10 ng/ml: IL-6 vs IL-1β: P = 0.007 (−0.8398, 95%CI [−1.374, −0.3059]). No difference between TNF-α and IL-6 was found at any concentration. IL-1β appeared to be the strongest inducer of FGF23 expression in chondrocytes.
Figure 1. IL-6, TNF-α and IL-1β upregulated FGF23 and downregulated SOX9 gene expression in HUM-iCell-s018 chondrocyte cells. Chondrocytes were treated with gradient concentrations of IL-6, TNF-α or IL-1β (0, 1, 5, 10 ng/ml) for 24 h and gene expression of FGF23 and SOX9 in chondrocytes was determined by qRT-PCR (a – d). Data are expressed with mean ± SD (a, c): *p < 0.05, **p < 0.01 and ***p < 0.001, the colour of * correspond with cytokine colour curve; (b, d): #p < 0.05: TNF-α vs IL-6; ^p < 0.05: TNF-α vs IL-1β; +p < 0.05 and ++p < 0.01: IL-6 vs IL-1β. All experiments were repeated three times independently.
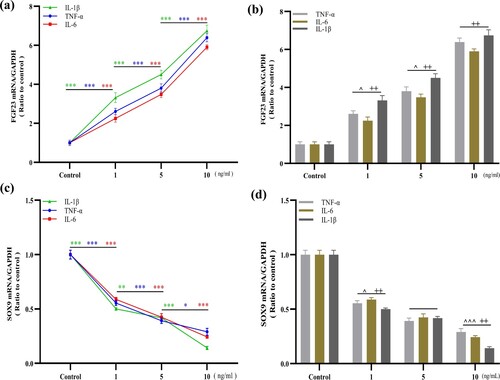
Figure 2. IL-6, TNF-α and IL-1β upregulated FGF23 and downregulated SOX9 protein expression in HUM-iCell-s018 chondrocyte cells. Chondrocytes were treated with gradient concentrations of IL-6, TNF-α or IL-1β (0, 1, 5, 10 ng/ml) for 24 h and protein expression of FGF23 and SOX9 in chondrocytes was determined by WB (a – e). Data are expressed with mean ± SD; (b – e) *p < 0.05, **p < 0.01 and ***p < 0.001. All experiments were repeated three times independently.
3.2. IL-6, TNF-α and IL-1β downregulate the SOX9 expression in chondrocytes
IL-6, TNF-α and IL-1β each downregulated the expression of SOX9 in chondrocytes in a dose-dependent manner ((c) and (a,d,e)) TNF-α: 0 vs 1 p < 0.001 (0.4451, 95%CI [0.3689, 0.5213]); 1 vs 5 p < 0.001 (0.1626, 95%CI [0.08638, 0.2388]); 5 vs 10 p = 0.012 (0.1013, 95%CI [0.02508, 0.1775]); IL-6: 0 vs 1 p < 0.001 (0.4113, 95%CI [0.3423, 0.4803]); 1 vs 5 p < 0.001 (0.1640, 95%CI [0.09505, 0.2330]); 5 vs 10 p < 0.001 (0.1815, 95%CI [0.1125, 0.2505]); IL-1β: 0 vs 1 p < 0.001 (0.4990, 95%CI [0.4416, 0.5564]); 1 vs 5 p = 0.0072 (0.08330, 95%CI [0.02589, 0.1407]); 5 vs 10 p < 0.001 (0.2762, 95%CI [0.2188, 0.3336]). We also compared the expression of SOX9 at identical concentrations treated with IL-6, TNF-α or IL-1β in order to identify the cytokine with the strongest inhibitory effect ((d)). At 1 and 10 ng/ml, both TNF-α and IL-6 exhibited stronger inhibition than IL-1β; 1 ng/ml: TNF-α vs IL-1β: P = 0.0278 (0.05390, 95%CI [0.007026, 0.1008]); IL-6 vs IL-1β: P = 0.0026 (0.08770, 95%CI [0.04083, 0.1346]); 10 ng/ml: IL-6 vs IL-1β: P = 0.0034 (0.1017, 95%CI [0.04455, 0.1588]); TNF-α VS IL-1β: P < 0.001 (0.1017, 95%CI [0.09235, 0.2066]). TNF-α and IL-6 seemed to have stronger inhibitory effects on SOX9 expression in chondrocytes.
4. Discussion
Studies have confirmed that unbalanced inflammatory reactions and oxidative stress caused by proinflammatory cytokines play vital roles in the pathogenesis of haemophilic cartilage degradation. Despite numerous studies on haemophilic cartilage destruction, much of the focus has been on chondrocyte apoptosis and ECM degradation, whereas little attention has been given to the interaction between proinflammatory cytokines and changes in the chondrocyte phenotype.
It is currently agreed that cartilage destruction in HA is induced by the combination of inflammation, joint bleeding and mechanical stress. Inflammation-induced cartilage destruction causes joint load disorders that increase vulnerability to mechanical stress. Additionally, inflammation induced synovium heterogeneous hyperplasia leads to disturbances in HA joint load conduction and abnormal mechanical stress. Hypertrophic synovium and abnormal mechanical stress may aggravate joint bleeding which decreases the viscoelasticity of the cartilage and increases the coefficient of friction of the joint surfaces, making them more susceptible to damage by mechanical stress. Cartilage destruction in OA is mainly due to mechanical stress and rheumatoid arthritis (RA) is mainly due to inflammatory factors, while HA is characterized by cartilage destruction due to both inflammatory and degenerative joint changes.
We found that IL-6, IL-1β and TNF-α each downregulated the expression of SOX9 in chondrocytes in a dose-dependent manner. Previous studies have reported that the overproduction of proinflammatory cytokines could induce excessive oxidative stress, which would downregulate the SOX9 expression in chondrocytes through the nuclear factor-κB (NF-κB) pathway [Citation17,Citation18]. This study presents the first documented relationship between IL-6, IL-1β and TNF-α and FGF23 expression in chondrocytes. The effects of proinflammatory cytokines on FGF23 expression have been extensively studied in chronic kidney disease (CKD) [Citation19,Citation20]. It has been shown that IL-6 increased FGF23 expression in osteoblast-like UMR106 cells through STAT3 phosphorylation, while TNF-α and IL-1β induced FGF23 expression in IDG-SW3 cells via NF-κB signalling [Citation21,Citation22]. NF-κB and STAT signalling pathways are also involved in the regulation of FGF23 expression in macrophages [Citation23]. Interestingly, both NF-κB and JAK/STAT pathways play crucial pathological roles in cartilage degradation [Citation24]. Hence, it's possible that IL-1β, TNF-α and IL-6 also up-regulate the expression of FGF23 in chondrocytes through the activation of NF-kB or/and JAK/STAT pathways.
To determine the independent effects of IL-6, IL-1β and TNF-α on the expression of FGF23 and SOX9 in chondrocytes, their activities at similar concentrations were compared. IL-1β was observed to be mainly involved in chondrocyte metabolism by stimulating FGF23 instead of SOX9, which is already well-documented. However, TNF-α and IL-6 were mainly involved in SOX9 regulation. Evidence obtained from existing literature focuses largely on the correlation between proinflammatory cytokines and SOX9, yet more attention should be given to the links between proinflammatory cytokines and FGF23.
Immunotherapy has been widely approved for the treatment of patients with RA, a condition in which proinflammatory cytokines such as IL-6, IL-1β and TNF-α play important pathological roles [Citation25–30]. TNF-α antagonists (infliximab, etanercept, adalimumab, golimumab and certolizumab pegol), IL-6 antagonists (siltuximab, olokizumab and clazakizumab), IL-1β antagonists (canakinumab), IL-6 receptor antagonists (tocilizumab and satralizumab) and IL-1β receptor antagonists (anakinra) have been widely used in RA treatment to delay disease progression. Reports have confirmed that immunotherapy could effectively reduce disease activity, decrease total joint score progression, slow the pace of joint destruction and improve function in RA patients. IL-6, IL-1β and TNF-α also play an important part in HA cartilage destruction pathogenesis, so immunotherapy may also useful for delaying progressive cartilage damage and improving symptoms of HA patients.
Although we found that IL-6, IL-1β and TNF-α could each alter the chondrocyte phenotype by regulating the expression of FGF23 and SOX9, which can be important causes of haemophilic cartilage degradation, our research did have some limitations. For example, we did not explore the mechanism between the proinflammatory cytokines which will be examined in future studies.
5. Conclusion
Our findings suggest that proinflammatory cytokines, IL-6, IL-1β and TNF-α, may be involved in haemophilic cartilage destruction pathogenesis by altering the chondrocyte phenotype. Modulation of FGF23 and SOX9 expression in the chondrocyte occur as IL-1β stimulates FGF23 expression, while TNF-α and IL-6 have regulatory effects on SOX9 expression.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801–1809.
- Zhu H, Meng Y, Tong P, et al. Pathological mechanism of joint destruction in haemophilic arthropathy. Mol Biol Rep. 2021;48(1):969–974.
- Srivastava A. Inflammation is key to hemophilic arthropathy. Blood. 2015;126(19):2175–2176.
- Razzaque MS. Interactions between FGF23 and vitamin D. Endocr Connect. 2022;11(10):e220239.
- Hasan M, Oster M, Reyer H, et al. Tissue-wide expression of genes related to vitamin D metabolism and FGF23 signaling following variable phosphorus intake in pigs. Metabolites. 2022;12(8):729.
- Roumpou A, Yavropoulou MP, Chronopoulos E, et al. Novel therapeutic agents for rare diseases of calcium and phosphate metabolism. Horm Metab Res. 2022;54(10):645–657.
- Bianchi A, Guibert M, Cailotto F, et al. Fibroblast Growth Factor 23 drives MMP13 expression in human osteoarthritic chondrocytes in a Klotho-independent manner. Osteoarthritis Cartilage. 2016;24(11):1961–1969.
- Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013;140(20):4129–4144.
- Jouan Y, Bouchemla Z, Bardèche-Trystram B, et al. Lin28a induces SOX9 and chondrocyte reprogramming via HMGA2 and blunts cartilage loss in mice. Sci Adv. 2022;8(34):eabn3106.
- Nishimura R, Hata K, Takahata Y, et al. Regulation of cartilage development and diseases by transcription factors. J Bone Metab. 2017;24(3):147–153.
- Green JD, Tollemar V, Dougherty M, et al. Multifaceted signaling regulators of chondrogenesis: implications in cartilage regeneration and tissue engineering. Genes Dis. 2015;2(4):307–327.
- Zheng L, Han Z, Luo D, et al. FGF23 and SOX9 expression in haemophilic cartilage: In vitro studies of the effects of iron. Haemophilia. 2022;28(6):1062–1068.
- Mammen J, Nair SC, Srivastava A. External quality assessment scheme for hemostasis in India. Semin Thromb Hemost. 2007;33:265–272.
- Payal V, Sharma P, Goyal V, et al. Clinical profile of hemophilia patients in Jodhpur region. Asian J Transfus Sci. 2016;10:101–104.
- Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125(13):2038–2044.
- Abshire T. Unraveling hemophilic arthropathy. Blood. 2011;117(8):2302–2303.
- Abu-Zaid A, Magzoub D, Aldehami MA, et al. The effect of iron supplementation on FGF23 in chronic kidney disease patients: a systematic review and time-response meta-analysis. Biol Trace Elem Res. 2021;199(12):4516–4524.
- Sikura KÉ, Potor L, Szerafin T, et al. Potential role of H-Ferritin in mitigating valvular mineralization. Arterioscler Thromb Vasc Biol. 2019;39(3):413–431.
- Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018;44:38–50.
- Rigoglou S, Papavassiliou AG. The NF-kappaB signalling pathway in osteoarthritis. Int J Biochem Cell Biol. 2013;45(11):2580–2584.
- Durlacher-Betzer K, Hassan A, Levi R, et al. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018;94(2):315–325.
- Ito N, Wijenayaka AR, Prideaux M, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol Cell Endocrinol. 2015;399:208–218.
- Han X, Li L, Yang J, et al. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS Lett. 2016;590(1):53–67.
- Nishimura R, Hata K, Takahata Y, et al. Role of signal transduction pathways and transcription factors in cartilage and joint diseases. Int J Mol Sci. 2020;21(4):1340.
- Rubbert-Roth A, Furst DE, Nebesky JM, et al. A review of recent advances using Tocilizumab in the treatment of rheumatic diseases. Rheum Ther. 2018;5(1):21–42.
- Ma X, Xu S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep. 2013;1(2):177–184.
- Alten R, Gomez-Reino J, Durez P, et al. Efficacy and safety of the human anti-IL-1β monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, Phase II, dose-finding study. BMC Musculoskelet Disor. 2011;12:153.
- LiverTox: Clinical and research information on drug-induced liver injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012.
- Radner H, Aletaha D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med Wochenschr. 2015;165(1-2):3–9.
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2(5):364–371.