
ABSTRACT
Objective
In the present study, two unrelated cases of Hb Q-Thailand heterozygosity unlinked with the (–α4.2/) α+-thalassemia deletion allele were identified by long-read single molecule real-time (SMRT) sequencing in southern China. The aim of this study was to report the hematological and molecular features as well as diagnostic aspects of the rare manifestation.
Methods
Hematological parameters and hemoglobin analysis results were recorded. A suspension array system for routine thalassemia genetic analysis and long-read SMRT sequencing were applied in parallel for thalassemia genotyping. Traditional methods, including Sanger sequencing, multiplex gap-polymerase chain reaction (gap-PCR) and multiplex ligation-dependent probe amplification (MLPA), were used together to confirm the thalassemia variants.
Results
Long-read SMRT sequencing was used to diagnose two Hb Q-Thailand heterozygous patients for whom the hemoglobin variant was unlinked to the (–α4.2/) allele for the first time. The hitherto undescribed genotypes were verified by traditional methods. Hematological parameters were compared with those of Hb Q-Thailand heterozygosity linked with the (–α4.2/) deletion allele in our study. For the positive control samples, long-read SMRT sequencing revealed a linkage relationship between the Hb Q-Thailand allele and the (–α4.2/) deletion allele.
Conclusions
Identification of the two patients confirms that the linkage relationship between the Hb Q-Thailand allele and the (−α4.2/) deletion allele is a common possibility but not a certainty. Remarkably, as it is superior to traditional methods, SMRT technology may eventually serve as a more comprehensive and precise method that holds promising prospects in clinical practice, especially for rare variants.
1. Introduction
Hemoglobinopathies, including thalassemia and hemoglobin variants, are widespread in Asia, especially in Southeast Asia and southern China. Hemoglobin variants are a group of hereditary hemoglobin diseases caused by mutations in globin genes that lead to structural changes in the hemoglobin molecule. To date, more than 1400 hemoglobin variants have been listed in the comprehensive hemoglobin variant database (http://globin.cse.psu.edu./hbvar/menu.html) [Citation1,Citation2]. Among those variants, Hb Q-Thailand [α74(EF3)Asp→His], caused by a missense mutation (GAC > CAC) at codon 74 of the α1-globin gene (HBA1) on chromosome 16p, has occasionally been reported. As one of the most common hemoglobin variants in southern China, Hb Q-Thailand is characterized by an association with a leftward single α2-globin gene (HBA2) deletion (–α4.2/) allele, and to date, no evidence of a single Hb Q-Thailand heterozygote unlinked with the (–α4.2/) allele has been accurately documented [Citation3,Citation4].
With the development of third-generation sequencing (TGS) methods for obtaining contiguous sequence information from long molecules, long-read single molecule real-time (SMRT) sequencing, the earliest commercial technical approach on the PacBio Sequel platform, has been evaluated as an alternate method for genotyping carriers of thalassemia [Citation5,Citation6]. In our study, using long-read SMRT sequencing for the genetic diagnosis of thalassemia, we detected two cases of heterozygous carriers of Hb Q-Thailand unlinked to the (–α4.2/) allele for the first time and subsequently validated the variants by a combination of traditional genotyping methods. Compared with other genotyping techniques, SMRT sequencing can detect variants more comprehensively and precisely and can provide additional information for the phasing of variant alleles, which is especially crucial for compound heterozygosity of recessive Mendelian disorders.
2. Materials and methods
2.1. Subjects and hematological analysis
Both patients originated from Guangdong Province, China. Case 1 is a 23-year-old female who was referred to our hospital for a routine prenatal examination at the 26th week of gestation and then underwent thalassemia screening. Case 2 is a 31-year-old man who attended our hospital for routine partner testing for thalassemia. These two cases were from unrelated families. Both patients agreed to participate in our study and signed informed consent forms. To serve as the positive controls, samples from twenty-seven Hb Q-Thailand heterozygous individuals (hereafter referred to as ‘PC samples’) with known linkages to the (–α4.2/) allele who had been diagnosed in recent years at our hospital were collected and examined simultaneously. All studies were approved by the Ethics Committee of Guangdong Women and Children Hospital. Blood samples were collected, and hematological parameters were determined using a Sysmex XN5000 automated hematology analyzer (Sysmex Corporation, Kobe, Japan). Hb analysis was conducted with an automated capillary 2 electrophoresis system (Sebia, France).
2.2. Suspension array-based routine thalassemia genotyping
Genomic DNA was extracted from peripheral blood leukocytes using the Lab-Aid 820 automation system (Zee San Biotech Company, Fujian, China). Twenty-three α- and β-thalassemia mutations common in southern Chinese individuals were routinely measured in our laboratory by a suspension array system, as previously described [Citation7].
2.3. Long-read SMRT sequencing
To evaluate whether SMRT sequencing might be suitable for the clinical genotypic analysis of thalassemia, the samples were also sent to an independent laboratory (Berry Genomics, Beijing, China) for parallel detection of thalassemia variants by long-read SMRT sequencing and data analysis. Briefly, multiplex long-range PCR was performed to generate specific amplicons that can simultaneously capture currently known indels (insertions and deletions) and single nucleotide variations (SNVs) in the α1-, α2- and β-globin genes. For construction of the presequencing library, amplicons were subjected to end repair and barcoded by ligation of adapters. The SMRTbell libraries were then prepared from the prelibrary. Primer annealing and polymerase binding to SMRTbell templates were performed, and sequencing was initiated on the PacBio Sequel System. Sequencing data analysis was carried out as previously published [Citation5,Citation6]. Integrative Genomics Viewer (IGV) was used to display alignments of variant and wild-type molecules.
2.4. Confirmation of thalassemia variants using traditional methods
To detect hemoglobin variants, the α- and β- globin genes were subjected to polymerase chain reaction (PCR) amplification, and then DNA Sanger sequencing of the amplified products was performed on an ABI PRISM 3130XL Analyzer (Applied Biosystems, USA). Three common deletion mutations of the α-globin gene cluster genotype, (– – SEA/), (–α3.7/) and (–α4.2/), were analyzed by multiplex gap-PCR according to the manufacturer’s instructions (Yaneng Bioscience, Shenzhen, China). Multiplex ligation-dependent probe amplification (MLPA) was used to confirm the α-globin gene cluster deletion mutation. Probes and reaction mixtures were purchased from MRC-Holland (SALSA MLPA kit P140 HBB, MRC-Holland, Amsterdam, the Netherlands). All techniques were performed according to standard procedures.
2.5. Statistical analysis
The hematological parameters of the PC samples are described using the mean and standard deviation.
3. Results
The hematological indices and genotypes are listed in . The hematological parameters showed that Case 1 was anemic with a hemoglobin level of 106 g/L. Decreased values of the red blood cell count (RBC), hematocrit (HCT), and ferritin (FER) were useful evidence of iron deficiency anemia. Both of the cases had a normal mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH). Hematological analysis of the PC samples showed that the hemoglobin level was within the normal range and borderline hypochromic according to the microcytic parameters. Further hemoglobin analysis showed that the electropherograms of the cases and PC samples were similar. For both cases (A), qualitative and quantitative estimation of Hb revealed abnormal fractions of 15.8% and 15.5%, while the levels among the PC samples (B and ) were higher (27.8 ± 2.9%). Similar to the electropherogram of the PC samples, the abnormal peak was observed at electrophoretic zone 7 for the cases, which overlapped with the hemoglobin F (Hb F) peak.
Figure 1. Hemoglobin analysis by using CE. (A) Similar results for the two patients in the present study. (B) The result of a representative PC sample is shown.
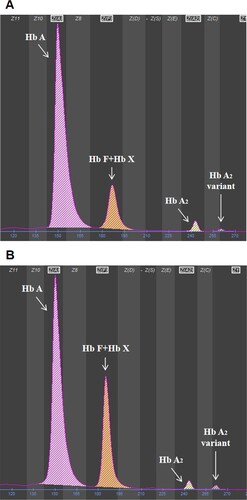
Table 1. Hematological and genotypic data of two patients and PC samples.
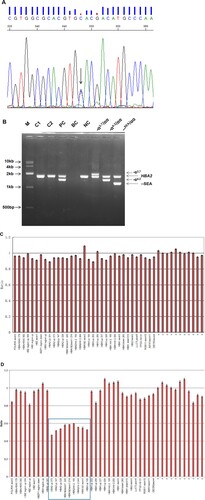
In both cases, no common α- and β-thalassemia mutations were found by the suspension-array system (data not shown). Long-read SMRT sequencing identified a heterozygous substitution (GAC > CAC) at codon 74 of the α1-globin gene (HBA1: c.223G > C) in all of the samples, corresponding to Hb Q-Thailand (). Surprisingly, neither of the two methods detected the (–α4.2/) deletion in the two cases, which was not in keeping with traditional views. Sanger sequencing confirmed that the codon 74 (G > C) substitution was present in both cases (A). The multiplex Gap-PCR showed that Case 1 and Case 2 were negative for all three common deletional mutations. No other α-globin gene cluster deletion mutation was detected by MLPA analysis in the two cases (B, C). A genotype result of –α4.2/αα from a representative PC sample was combined with the results of multiplex gap-PCR and MLPA analysis (B, D). Summarizing these measured results, the patients were diagnosed with heterozygous Hb Q-Thailand unlinked to the (–α4.2/) deletion allele.
Figure 2. IGV plots of HBA1 and HBA2 gene variants using long-read SMRT sequencing. The relative positions of the HBA2 and HBA1 genes on chromosome 16 are indicated by blue boxes. For the allelic reads, the yellow and blue regions represent the alignment of individual molecules, purple dots indicate sequencing errors, and the vertical colored lines indicate nucleotides A (green), T (red), C (blue), and G (orange) discordant with alignment to the reference sequence. (A) The same result was obtained for the two patients in the present study. The arrow indicates the G > C heterozygous substitution at codon 74 of the HBA1 gene, which corresponds to Hb Q-Thailand. (B) As a control, a representative result from a PC sample is shown. In addition to missing the heterozygous substitution, the deleted region is indicated by the gray box. The two variants are identified in a cis configuration.
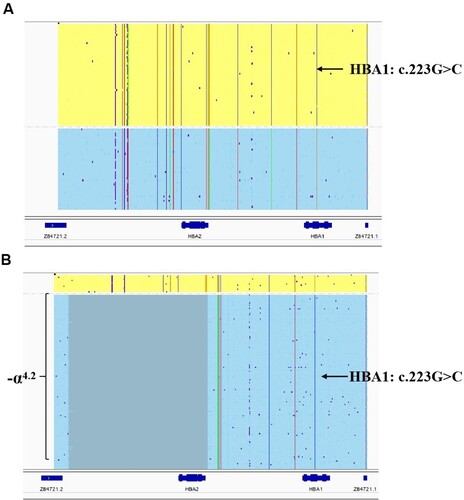
Figure 3. The genotyping results of the two patients by traditional methods. Sanger sequencing results. The arrow indicates the G > C heterozygous substitution at codon 74 of the HBA1 gene. (B) Genotyping for three common deletional mutations of α-globin gene clusters by multiplex gap-PCR. M: DL10000 DNA Marker; C1: Case 1; C2: Case 2; PC: – α4.2/αcodon74(GAC > CAC)α; BC: blank control; NC: αα/αα. (C) and (D): The MLPA results after normalization with a normal individual. The relative position of the probes in the α-globin gene cluster is indicated on the horizontal axis. The vertical axis shows the normalized probe ratio. (D) As a control, a representative result from a PC sample is shown. The blue box indicates the position of the (–α4.2/) deletion.
4. Discussion
Hb Q-Thailand is a slow-moving abnormal hemoglobin that is characterized by a substitution of aspartic acid with histidine at the 74th position of the normal α1-globin chain. This hemoglobin shows normal oxygen affinity, a normal Bohr effect and normal cooperativity because the substituted amino acid is nonfunctional [Citation8]. It was first observed in 1958 by Vella et al. in Singapore and first discovered in the Chinese population in 1983 by Zeng et al. [Citation9,Citation10]. Hb Q-Thailand has a high detection rate in southern China and Southeast Asian countries, where it occurs mostly in a heterozygous or compound heterozygous state with α0-thalassemia, causing Hb Q-H disease [Citation3,Citation4,Citation8,Citation11–13]. Although Hb Q-Thailand has been found in various populations, the present study is the first to report a negative result for DNA analysis of the (–α4.2/) deletion.
The hematological findings of PC samples were in keeping with those of heterozygous individuals in a previous survey [Citation3,Citation4]. The heterozygous form is usually clinically asymptomatic and has normal or slight hypochromic microcytosis because of the linked (–α4.2/) deletion allele. The quantitative estimation of Hb F and Hb Q in total is approximately 30% by the CE method because the Hb Q-Thailand fraction is unseparated from the Hb F fraction on the CE electropherogram. In the present study, based on the lack of the (–α4.2/) deletion allele, the two cases did not show hypochromic microcytic parameters and had higher MCV and MCH values as compared to those of PC samples. Additionally, there are two normal α-globin genes for those with Hb Q-Thailand linked to the (–α4.2/) deletion allele, but in the two cases presented, there are three normal α-globin genes. The levels of Hb A and Hb A2 are presented relative to Hb F & Hb Q-Thailand. The two cases showed a significantly lower percentage of Hb F plus Hb Q (A, ), which is probably due to the higher percentages of Hb A plus Hb A2.
Although Hb Q-Thailand heterozygosity was once reported to be unassociated with the (–α4.2/) deletion allele, subsequently conflicting results (the heterozygous mutation detected by Sanger sequencing contradicts the presence of the – –SEA/αα deletion) were not investigated in depth, which made the result unreliable [Citation14]. In our study, the Hb Q-Thailand variant detected by long-read SMRT sequencing was confirmed by Sanger sequencing, and multiplex gap-PCR, and the MLPA method verified that the (–α4.2/) deletion allele was undetected. Combining these methods, we ruled out the possibility that the existence of other mutations in this region of the designed primer caused a dropout of the αα allele in the suspension array method. During the routine diagnostic procedures for our two patients, if SMRT sequencing had not been applied, then the Hb Q-Thailand variant would not have been taken into consideration, and the (–α4.2/) deletion would not have been detected. Subsequently, the high percentages of Hb F would have logically indicated other rare variants. The simultaneous application of the suspension array-based routine assay and SMRT sequencing ensured the accuracy of the results and revealed that the Hb Q-Thailand variant was not necessarily linked to the (–α4.2/) deletion allele, which was different from previous reports ().
Currently, most thalassemia diagnostic strategies involve a combination of different targeted PCR-based methodologies to identify the most prevalent and known pathogenic variants. More recently, next-generation sequencing (NGS) has emerged as an alternative, more comprehensive and effective molecular approach for thalassemia gene screening [Citation15]. However, the short read lengths of NGS pose a limitation for the identification of structural variants, sequencing repetitive regions, phasing of alleles and distinguishing highly homologous genomic regions, which may have a significant impact on the need for fast field testing and widespread clinical practice. With the advantage of TGS methods, various studies have demonstrated the power of targeted long-read sequencing to compensate for the diagnostic gap of prevailing NGS [Citation16–18]. SMRT technology with long-range PCR provides a comprehensive and valuable genetic analysis and enables the identification of common, rare and unknown variants with full-length coverage of alleles in a single assay format in thalassemia genetic carrier screening [Citation5,Citation6]. In addition, SMRT technology can also discriminate compound heterozygotes in a cis or trans configuration. When two variants are detected in the same gene, especially for compound heterozygosity underlying a recessive disorder, it is critical to confirm the configuration to clarify the exact carrier state for diagnostic purposes. An individual with a single allele carrying two variants in a cis configuration is still a carrier [Citation19,Citation20]. As it is superior to testing the parents of the proband for confirmation, SMRT technology enables the direct detection and phasing of variants simultaneously. Whether in postnatal or prenatal cases, when compound heterozygous individuals show a mutation-unrelated phenotype or there are no genotypes available from the parents, SMRT technology is the best choice for resolving the confounding genotype-phenotype correlations. Although it has been described in many Asian populations, the linkage relationship between the Hb Q-Thailand variant and the (–α4.2/) deletion has only been researched once by analysis of the α-globin gene haplotype [Citation3]. In our study, the compound heterozygote used as the positive control was in a cis configuration. This linkage relationship was first exhibited by SMRT technology, which is a simpler and more intuitive method than previous analyses (). At present, all the methods for thalassemia genetic analysis have some limitations. There are a few mutations that SMRT cannot detect, but the traditional test can detect. When SMRT-negative cases have positive hematology screening in diagnostic settings, other traditional tests need to be considered. Due to cost limitations, SMRT technology is mainly used as a supplemental platform for the detection of rare thalassemia mutations in clinical diagnostic settings. With the development of technology and cost reduction, it is believed that SMRT technology will be applied in routine thalassemia genetic carrier screening. Due to the optimization of testing platforms, more rare thalassemia mutations may be detected. For rare mutations with unknown pathogenicity, hematological data must be studied to judge whether there is a thalassemia phenotype. Analysis of family members with the same rare mutation by hematological and genotypic data would provide more evidence, which may be helpful in genetic counseling.
In our study, for the first time, we identified two Hb Q-Thailand heterozygous cases unlinked with the (–α4.2/) deletion allele. The application of SMRT technology showed improved efficiency and accuracy over routine traditional methods. These advantages make SMRT technology a promising prospect for clinical application. In conclusion, this study is an important complement to previous reports and could be useful for promoting accurate genetic diagnosis, appropriate genetic counseling and prenatal diagnostic services in this population.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data sources from which the results of this study were generated can be shared if requested.
Additional information
Funding
References
- Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42(Database issue):D1063–D1069. doi:10.1093/nar/gkt911.
- Giardine BM, Joly P, Pissard S, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2021;49(D1):D1192–D1196. doi:10.1093/nar/gkaa959.
- Singsanan S, Karnpean R, Fucharoen G, et al. Hemoglobin Q-Thailand related disorders: origin, molecular, hematological and diagnostic aspects. Blood Cells Mol Dis. 2010;45(3):210–214. doi:10.1016/j.bcmd.2010.06.001.
- Xu A, Chen W, Xie W, et al. Hemoglobin variants in southern China: results obtained during the measurement of glycated hemoglobin in a large population. Clin Chem Lab Med. 2020;59(1):227–232. doi:10.1515/cclm-2020-0767.
- Xu L, Mao A, Liu H, et al. Long-Molecule sequencing: a new approach for identification of clinically significant DNA variants in α-Thalassemia and β-ThalassemiaCarriers. JMolDiagn. 2020;22(8):1087–1095. doi:10.1016/j.jmoldx.2020.05.004.
- Liang Q, Gu W, Chen P, et al. A more universal approach to Comprehensive Analysis of Thalassemia Alleles (CATSA). J Mol Diagn. 2021;23(9):1195–1204. doi:10.1016/j.jmoldx.2021.06.008.
- Luo MY, Hu TT, Wang JC, et al. Application of a suspension array based on barcoded magnetic beads in thalassemia gene diagnosis. J Trop Med. 2016;16(7):832–835.
- Leung KF, Ma ES, Chan AY, et al. Clinical phenotype of haemoglobin Q-H disease. J Clin Pathol. 2004;57(1):81–82. doi:10.1136/jcp.57.1.81.
- Vella F, Wells RH, Ager JA, et al. A haemoglobinopathy involving haemoglobin H and a new (Q) haemoglobin. Br Med J. 1958;1(5073):752–755. doi:10.1136/bmj.1.5073.752.
- Zeng YT, Huang SZ, Zhou XD, et al. A study on one family with hemoglobin Q Thailand (a2 74(EF3)Asp—His) found in China. Acta Biochim Biophys Sin, 1983; 15(5): 423-432.
- Panyasai S, Fucharoen G, Fucharoen S. Hemoglobin variants in Northern Thailand: prevalence, heterogeneity and molecular characteristics. Genet Test Mol Biomarkers. 2016;20(1):37–43. doi:10.1089/gtmb.2015.0182.
- Li YQ, Chen ZZ, Liang L, et al. Analysis of the phenotype-genotype relationship of hemoglobin Q-Thailand in Guangxi. Chin J Med Genet. 2016;33(2):164–168. doi:10.3760/cma.j.issn.1003-9406.2016.02.008.
- Zhong ZY, Chen JH, Guan ZY, et al. Phenotypic and genotypic analysis of 45 cases with Hemoglobin Q-Thailand. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018;35(5):723–726. doi:10.3760/cma.j.issn.1003-9406.2018.05.024.
- Zhang Q, Fan X, Wei Y, et al. Genetic analysis on HB Q-Thailand in Nanning city of Guangxi. Lab Med Clin. 2014;11(20):2803–2804+2807.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with Hemoglobinopathies. EBioMedicine. 2017;23:150–159. doi:10.1016/j.ebiom.2017.08.015.
- Wenger AM, Peluso P, Rowell WJ, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37(10):1155–1162. doi:10.1038/s41587-019-0217-9.
- Mantere T, Kersten S, Hoischen A. Long-Read sequencing emerging in medical genetics. Front Genet. 2019; 10: 426. doi:10.3389/fgene.2019.00426
- Midha MK, Wu M, Chiu KP. Long-read sequencing in deciphering human genetics to a greater depth. Hum Genet. 2019;138(11–12):1201–1215. doi:10.1007/s00439-019-02064-y.
- Veyradier A, Fressinaud E, Goudemand J. Compound heterozygosity or heterozygosity with two mutations in cis on the same allele? A rebuttal to the letter to the editors: Koessler et al. Von Willebrand disease caused by compound heterozygosity for p.R854Q and p.R760C: diagnostic and therapeutic implications. Haemophilia. 2011; 17(5): e832–e833. doi:10.1111/j.1365-2516.2011.02531.x
- Li J, Jiang F, Zhen L, et al. A β-Thalassemia trait with two mutations in Cis in a Chinese family. Hemoglobin. 2019;43(4–5):289–291. doi:10.1080/03630269.2019.1686011.