
ABSTRACT
Background:
Sickle Cell Anemia (SCA) is the most common genetic disease worldwide caused by a single mutation in the gene HBB. The disease severity is very variable and depends on many factors. We evaluated the clinical and biological profile of sickle cell anemia children in rural Central Africa.
Methods:
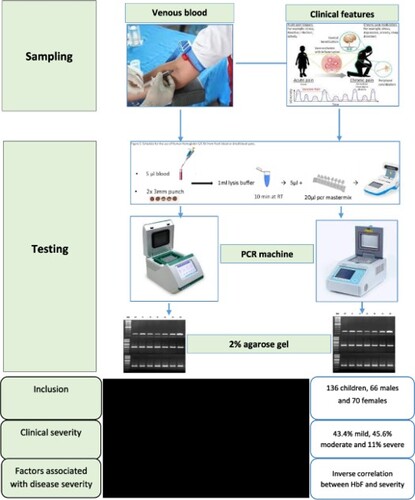
This cross-sectional study was conducted in the Hôpital Saint Luc de Kisantu, located 120 km away from Kinshasa-DR Congo in an area of 35 km around Kisantu with a population of roughly 80 000 individuals. We included SCA patients aged 6 months to 18 years. We collected clinical and hematological data. The SCA scoring system proposed by Adegoke et al. in 2013 was applied to determine the disease severity. We searched for factors associated to the disease severity.
Results:
This study included 136 patients, 66 males and 70 females (sex-ratio M/F 0.94). The mean severity score was 8.21 ± 5.30 (ranges 0–23). Fifty-nine (43.4%) children had mild disease, 62 (45.6%) moderate and 15 (11%) severe disease. Girls had higher levels of HbF than boys (p = 0.003). An inverse correlation was observed between fetal hemoglobin and the disease severity (p = 0.005, r −0.239, IC95% −6.139; −1.469). Some factors such age influence the occurrence of certain chronic complications such as avascular bone necrosis.
Conclusion:
In conclusion, the disease severity of SCA depends on multiple factors. In this study, fetal hemoglobin was the main modulator of the disease severity. These data may also serve as a baseline to initiate HU treatment in this setting.
GRAPHICAL ABSTRACT
Introduction
Sickle Cell Anemia (SCA) is one of the most common severe genetic disorders worldwide [Citation1,Citation2]. Its prevalence is particularly high in sub-Saharan countries, especially in the Democratic Republic of Congo (DRC) where 1.4–2.3% of infants are born with SCA. Thus, the DRC ranks third among the countries with the highest number of affected patients in the world, after Nigeria and India [Citation3–5]. An estimated 50-90% of SCA children die before age of 5 years [Citation6–8]. The sickle hemoglobin variant (HbS) results from a single mutation, the substitution of glutamic acid by valine at position 7 of β globin (E7 V) [Citation9]. HbS polymerizes in the red blood cells under deoxygenated conditions. This results in a chain of events leading to acute and chronic manifestations due to a combination of vaso-occlusion, chronic hemolysis, inflammation, and endothelial dysfunction. The clinical features and the severity of the disease vary considerably from one SCA patient to another but also over time in an individual patient [Citation10,Citation11]. Assessing clinical severity is a means to identify a high-risk group who needs close follow-up and may be eligible for hydroxyurea (HU) treatment. Several scores evaluating the severity of the disease in children have been proposed. Most of these includes both clinical manifestations and biological or laboratory measurements [Citation12–15]. The main difference resides in the number and the type of features included as well as in the number of severity categories. The scores also vary in their user-friendliness. In that particular regard, the score developed by Adegoke in Nigeria appears to be most suitable to the setting in a rural environment in Central Africa, because this score relies on six clinical parameters that are easy to assess and two standard biological parameters (packed cell volume and white blood cells). In this score, 15 parameters were assessed to reflect each patient’s present state, their state during the previous 12 months, and lifetime complications. Items were scored according to the frequency of occurrence and severity, with scores ranging from 1 to 5, as shown in supplementary Table 1. For acute life-threatening events and neurological complications, higher scores were assigned: cerebrovascular disease (CVD) was assigned a score of 5, acute chest syndrome (ACS) was assigned a score of 3, and avascular bone necrosis (AVN) was assigned the score of 3 because even though it is a severe complication of SCA, it does not immediately lead to acute life-threatening events. This score has been applied in several studies from Sub-Saharan Africa [Citation16–20].
The clinical severity is influenced by multiple factors such as the level of fetal hemoglobin (HbF), including genetics, age, sex, nutritional and socioeconomic state, access to medical care and comorbidities such as thalassemia and malaria. SCA children in rural areas live in a true vicious circle where malnutrition leads to an increase in both acute and chronic complications, triggering increased health expenses and thus aggravation of poverty, ultimately exacerbating the malnutrition [Citation21–23]. The recent introduction of HU to ameliorate SCA disease severity is promising both in children and adults. However, its use is still limited in sub-Sahara Africa.
This study aims to determine the clinical and biological features of SCA children in a rural area of central Africa and evaluate the factors associated to the disease severity. Since these SCA children previously only receive the basic treatment of penicillin and folic acid, these data may serve as a baseline to initiate HU treatment in this setting.
Methods
Recruitment and data collection
This is a cross-sectional study conducted in Kisantu Saint Luc Hospital (KSLH), in a rural area located 120km west from Kinshasa the capital. KSLH is the major referral hospital in the region, following around 250 adult and children SCA patients. During a period of 12 months, from September2017 to end of August 2018, we included patients aged 6 months to 18 years meeting the following inclusion criteria: confirmed SCA by both capillary hemoglobin electrophoresis and DNA testing. The patients had also to be in steady state, defined by the absence of painful crises and infections during the past four weeks, the absence of blood transfusion in the past four months. Parents or legal guardians had to provide a signed consent form.
We recorded the following variables: age, gender, number of hospitalizations (in days), transfusions and painful crises during the previous year, and past occurrence of acute chest syndrome, avascular bone necrosis, priapism, clinical signs of cerebral infarct and acute splenic or hepatic sequestration. We obtained a full blood cells count (red blood cells (RBC), white blood cells (WBC), platelets and reticulocytes). Biochemical analyses included lactate dehydrogenase (LDH), bilirubin (Bili), serum creatinine (Crea), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) (Laboratory of Biochemistry and Hematology, Faculty of Pharmaceutical Sciences of the University of Kinshasa (UNIKIN)).
Hemoglobin electrophoresis was performed using the automate Minicap (Sebia, Phoresis Rel 8.6.3.), DNA was extracted by the salting out method and mutation analysis for the SCA mutation (E7 V) were performed in the Laboratory of Human Genetics at UNIKIN. A restriction fragment length polymorphism (RFLP) test for SCA was performed, using the Ddel restriction enzyme to differentiate between the mutant allele (S) and wild-type allele (A) of a PCR product spanning the SCA mutation site [Citation24].
We applied the SCA scoring system proposed by Adegoke et al. [Citation15] to determine the disease severity. This scoring system contains 15 parameters that assess patient state during the previous 12 months and lifetime complications. Items were scored depending on the frequency of occurrence and the severity of the event. A score between 0 and 5 was assigned to items (Supplementary Table 1). The total score equaled the sum of scores obtained by individual items and ranged from 0 to 34. This allowed to distinguish 3 categories of disease severity: smild (total score < 8), moderate (8–17), and severe (>17).
Ethical committee
The study was approved by the ethical committee of the school of public health of the University of Kinshasa (Approval reference: ESP/CE/079/2016), DRC. We provided necessary information to the family (parent or guardian) prior to the written consent. Age-appropriate assent was obtained from children. Information sheets and consent forms were written in two languages in Lingala (local language) and in French. Survey forms and sample tubes were labeled with unique patient identification codes to protect privacy.
Statistical analyses
We collected data on an Excel sheet in Microsoft Windows 2010 and imported and analyzed with IBM SPSS Statistics 25.0 software. Qualitative data (sex, nutritional status, severity score, molecular testing result) were presented as proportions whereas quantitative data (age, weight, height, biological parameters) were presented using mean and/or median and standard deviation. Columns and Bars were used to provide clear data visualization. Anova with a Bonferroni post-hoc test was used to compare the values between quantitative variable and qualitative one with more than two groups; Student tests were used to compare means and Chi-square and Fischer tests compared proportions. Mann Whitney-U test compared medians when the data did not follow a normal distribution. Logistic regression allowed us to establish associations between variables. The level of statistical significance was defined by a p-value less than 0.05.
Results
Description of the population
We recruited 136 children with homozygous form of SCA (SS children) in an area of 35 km around the hospital, 66 males and 70 females (sex-ratio M/F 0.94). Age ranged from 6 months to 18 years, with mean age (±standard deviation) 9.64 ± 4.55 years: males 9.0 ± 4.7 years and females 10.2 ± 4.3 years (p = 0.112) (). Thirty-one children (22.8%) were in preschool-age (1-5years), 49 (36%) were in school-age (6-10years) and 56 (41.2%) were adolescents (10-18 years) ().
Table 1. Description of the population (n = all patients = 136).
Table 2. Gender with clinical and biological features.
Clinical variables and disease severity
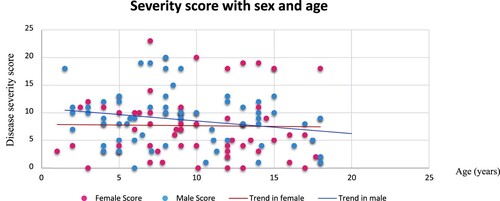
The mean severity score was 8.21 ± 5.30, ranging from 0 to 23. Fifty-nine (43.4%) children had mild disease, 62 (45.6%) moderate and 15 (11%) severe disease. The disease severity tended to be higher in males (8.77 ± 4.74) compared to females (7.67 ± 5.70), but this was not statistically significant (p = 0.225) (). Anova with Bonferroni post-hoc showed that children in school-age (6-10years) have a significantly higher severity score as compared to adolescents (Anova p = 0.016, Bonferroni p = 0.013). In males, the severity score showed a decrease with age, but this did not reach the statistical significance threshold (). Chronic complications were observed in 13/136 (9.6%) children including stroke in 4 children who were all males (2.94%), avascular bone necrosis in 7 children (5.15%), and leg ulcers in 2 children (1.47%); two children presented acute chest syndrome (1.47%). These complications added much weight to the severity score (Supplementary table 4). After logistic regression, stroke was not associated to any variables (supplementary table 2); but avascular bone necrosis was associated with age (OR 2.274, p = 0.045) (Supplementary table 3).
Figure 1. Disease severity with age and sex.
Biological data
Biological data showed normocytic normochromic anemia with high reticulocytes count and signs of hemolysis ( and ). Direct bilirubin and creatinine levels were significantly different between girls and boys, with higher levels in boys (). Mean HbF levels were 7.46%±1.86 (range 0-33.1), and were significantly higher in girls (10.73 ± 7.69%) compared to boys (8.64 ± 4.91%) (p = 0.003) (). We observed an inverse correlation between fetal hemoglobin and the disease severity score (p = 0.005, r −0.239, IC95% −6.139; −1.469) (). Moreover, fetal hemoglobin levels tended to decrease with age (). Biological factors not included in the severity score that were associated with disease severity include neutrophil count (higher in moderate versus severe disease, with p = 0.028) (Supplementary table 5) and HbF levels (inversely correlated with disease severity) (, supplementary table 5, ).
Figure 2. HbF levels with age and sex.
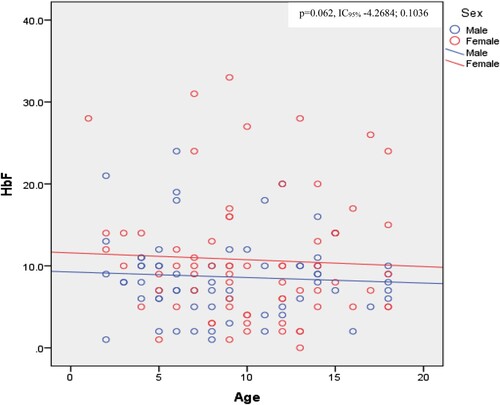
Figure 3. Disease severity with HbF levels.
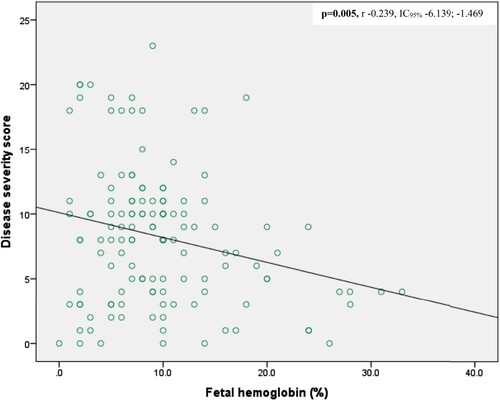
Table 3. Factors associated to disease severity.
Discussion
Studies of SCA clinical and biological profile in the Central Africa, especially in rural area are very scarce. First, since there is no national SCA neonatal screening program, only children presenting acute clinical event or chronic complications are ascertained. Second, given the high mortality in SCA, children with most severe presentation are under-represented in this cohort. In addition, although the consent form was written in French and Lingala, some parents were unable to read and understand the content. This fact highlights the complexity of working in a rural environment in Africa, where poverty and multilingualism have taken up residence. The disease severity of SCA varies from a patient to another, and in the same patient over the time [Citation10,Citation11]. That severity is depending on many factors related to the individual, the clinical parameters and biological variables. In a rural area, the clinical and biological profile may depend on certain factors such as the lack of quality care which reduces life expectancy; but also malnutrition and iron deficiency which can aggravate anemia. A comparative study in an urban environment could better illustrate the difference between the two environments.
Several clinical scores exist in the literature to assess disease severity of SCA [Citation12–15]. Among them, the Adegoke score seems to be the most suitable in our setting of a low-income country, especially in rural area. In rural hospitals, indeed, the technical platform is limited, and also people live in deep poverty so that they cannot afford some investigations. The Adegoke score appears therefore to be the most affordable since it relies on six clinical parameters that are easy to assess and two standard biological parameters (packed cell volume and white blood cells). In contrast, the other scores require some more investigations like MRI, Transcranial Doppler ultrasound, CT Scan, X-Ray. Regarding Adegoke score, some clinical parameters such as stroke, acute chest syndrome, avascular bone necrosis and leg ulcers contribute much to the score and allow to discriminate the different severity, because only individuals with severe form of the disease have presented those complications. Overall, parameters proposed by Adegoke score are not age and sex dependent, so that the score can be used either in pediatrics or in adult patients. Patients with a severe score showed a low neutrophil count, which is contrary to what is expected. We did not find a clear explanation for this. We hypothesized that the small study population size may be the cause. In addition, we did not find a significant difference between disease severity and hemolytic parameters such as LDH and bilirubin. We hypothesized that the hemolysis was high overall and that there may be other comorbid factors such as malaria that could explain this hemolysis. Unfortunately, we did not evaluate this in the present study. We noticed a negative correlation between fetal hemoglobin and disease severity (p = 0.005, r −0.239, IC95% −6.139; −1.469) (). The level of fetal hemoglobin is also dependent of sex; we observed a significant higher level in female (10.73 ± 7.69) compared to the male (8.64 ± 4.91) (p = 0.003) (). Fetal hemoglobin and fetal cell count are influenced by several genetic variants. Craig et al found two factors (beta thalassemia and Xmn I-G gamma site) on chromosome 11p. In addition to the two previous factors, Garner et al described a trans-acting locus controlling HbF and fetal cell production mapped to chromosome 6q23 [Citation25–28]. The fact that HbF is high in female than in male is not clearly explained, but was hypothesized to be related to polymorphisms in a locus located on Xp22.2. In fact, Dover at al 1992 observed that F-cell count was significantly higher in females than in males. It is known that the level of F-cell is related to HbF levels [Citation25,Citation26]. We thus confirm that the fetal hemoglobin is a main modulator of disease severity, also in our study [Citation1,Citation25]. Although the access to HU treatment is a major concern in a rural African setting due to poverty, studies in high-income countries show that socioeconomic factors, availability of HU and adherence to therapy remain barriers to this treatment also in other parts of the world [Citation29,Citation30]. In conclusion the fetal hemoglobin remains the main modulator of the disease severity. Some factors such age influence the occurrence of certain chronic complications such as avascular bone necrosis. We need therefore to look for this complication and treat it in old children. However, our study has some limitations. The first one is the small study population with a low power in some statistical tests. The second one is the fact that this is a cross-sectional study and not longitudinal study. Therefore, we were unable to determine the childhood mortality due to SCA in a rural environment in Central Africa.
Supplemental Material
Download MS Word (33.9 KB)Acknowledgements
We are grateful to The parents of SCA children who accepted to participate in this study. All the staff of pediatrics service of Kisantu Saint Luc Hospital. Cathy Songo, technician at the Center for Human Genetics, University of Kinshasa. Authors contributions: Data collection: Gloire Mbayabo. Drafting: Gloire Mbayabo, Mamy Ngole Zita, Paul Lumbala Kabuyi, Koenraad Devriendt, Chris Van Geet, Prosper Tshilobo Lukusa. Laboratory diagnosis of sickle cell disease: Gloire Mbayabo, Mamy Ngole. Conception and design of the study, review of the manuscript: Gloire Mbayabo, Mamy Ngole Zita, Paul Lumbala Kabuyi, Aimé Lumaka, Valerie Race, Gert Matthijs, Tite Mikobi Minga, Koenraad Devriendt, Prosper Tshilobo Lukusa, Chris Van Geet. All authors revised and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi:10.1016/S0140-6736(10)61029-X.
- Weatherall D, Akinyanju O, Fucharoen S, et al Chapter 34. inherited disorders of hemoglobin. In Disease control priorities in developing countries (2nd Ed). The International Bank for Reconstruction and Development / The World Bank 2006;663–680.
- Agasa B, Bosunga K, Opara A, et al. Prevalence of sickle cell disease in a northeastern region of the Democratic Republic of Congo: What impact on transfusion policy? Transfus Med. 2010;20(1):62–65.
- Tshilolo L, Kafando E, Sawadogo M, et al. Neonatal screening and clinical care programmes for sickle cell disorders in sub-Saharan Africa: Lessons from pilot studies. Public Health. 2008;122(9):933–941. doi:10.1016/j.puhe.2007.12.005.
- Tshilolo L, Aissi LM, Lukusa D, et al. Neonatal screening for sickle cell anaemia in the Democratic Republic of the Congo: Experience from a pioneer project on 31 204 newborns. J Clin Pathol. 2009;62(1):35–38. doi:10.1136/jcp.2008.058958.
- Grosse SD, Odame I, Atrash HK, et al. Sickle cell disease in Africa. Am J Prev Med. 2011;41(6):S398398–S39S405. doi:10.1016/j.amepre.2011.09.013.
- Kumar AA, Chunda-Liyoka C, Hennek JW, et al. Evaluation of a density-based rapid diagnostic test for sickle cell disease in a clinical setting in Zambia. PLoS One. 2014;9(12):1–24.
- Piel FB, Hay SI, Gupta S, et al. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7). e1001484. doi:10.1371/journal.pmed.100148.
- Inusa BPD, Hsu LL, Kohli N, et al. Sickle cell disease—genetics, pathophysiology, clinical presentation and treatment. Int J Neonatal Screen. 2019;5(2):20. doi:10.3390/ijns5020020.
- Ballas SK, Kesen MR, Goldberg MF, et al. Beyond the definitions of the phenotypic complications of sickle cell disease: An update on management. Sci World J. 2012;: 94953. doi:10.1100/2012/949535.
- Makani J, Ofori-Acquah SF, Nnodu O, et al. Sickle cell disease: New opportunities and challenges in Africa. Sci World J. 2013: 1–16. doi:10.1155/2013/193252.
- El-hazmi MAF. Clinical and haematological diversity of sickle cell disease in Saudi children. J Trop Pediatr. 1992;38(3):106–112. https://pubmed.ncbi.nlm.nih.gov/1380566/
- Sebastiani P, Nolan VG, Baldwin CT, Abad-Grau MM, Wang L, Adewoye AH, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110(7):2727–2735. doi:10.1182/blood-2007-04-084921
- Van Den Tweel XW, Van Der Lee JH, Heijboer H, et al. Development and validation of a pediatric severity index for sickle cell patients. Am J Hematol. 2010;85(10):746–751. doi:10.1002/ajh.21846.
- Samuel AA, Peter Kuti B. J Appl Hematol. 2013;4:58–64.
- Antwi-Boasiako C, Ekem I, Abdul-Rahman M, et al. Hematological parameters in Ghanaian sickle cell disease patients. J Blood Med. 2018;Volume 9:203–209. doi:10.2147/JBM.S169872.
- Uche E, Adelekan O, Akinbami A, et al. Serum homocysteine and disease severity in sickle cell anemia patients in Lagos. J Blood Med. 2019;10:127–134. doi:10.2147/JBM.S198316.
- Oladimeji OI, Adeodu OO, Onakpoya OH, et al. Prevalence of ocular abnormalities in relation to sickle cell disease severity among children in South-western, Nigeria. Eur J Ophthalmol. 2021;31(5):2659–2665. doi:10.1177/1120672120957615.
- Ezenwosu O, Chukwu B, Ezenwosu I, et al. Clinical depression in children and adolescents with sickle cell anaemia: influencing factors in a resource-limited setting. BMC Pediatr. 2021;21(1):1–8. doi:10.1186/S12887-020-02457-3.
- Okocha CE, Manafa PO, Igwe CN, et al. Adiponectin and disease severity in sickle cell anemia patients attending a tertiary health institution in Nnewi, Southeast Nigeria. Front Genet. 2022;13(February):1–5.
- Hyacinth HI, Gee BE, Hibbert JM. The role of nutrition in sickle cell disease. Nutr Metab Insights. 2010;3:NMI.S5048.
- Reid M. Nutrition and sickle cell disease. Comptes Rendus Biol. 2013;336(3):159–163. doi:10.1016/j.crvi.2012.09.007.
- Mandese V, Marotti F, Bedetti L, et al. Effects of nutritional intake on disease severity in children with sickle cell disease. Nutr J. 2016;15(1):1–6. doi:10.1186/s12937-016-0159-8.
- Mikobi TM, Lukusa Tshilobo P, Aloni MN, et al. Clinical phenotypes and the biological parameters of Congolese patients suffering from sickle cell anemia: A first report from Central Africa. J Clin Lab Anal. 2017;31(6):1–6.
- Carrocini GC de S, Zamaro PJA, Bonini-Domingos CR. What influences Hb fetal production in adulthood? Rev Bras Hematol Hemoter. 2011;33(3):231–236. doi:10.5581/1516-8484.20110059.
- Dover GJ, Smith KD, Chang YC, et al. Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2. Blood. 1992;80(3):816–824. doi:10.1182/blood.V80.3.816.816.
- Green NS, Ender KL, Pashankar F, et al. Candidate sequence variants and fetal hemoglobin in children with sickle cell disease treated with hydroxyurea. PLoS One. 2013;8(2):1–7.
- Garner C, Mitchell J, Hatzis T, et al. Haplotype mapping of a major quantitative-trait locus for fetal hemoglobin production, on chromosome 6q23. Am J Hum Genet. 1998;62(6):1468–1474. doi:10.1086/301859.
- Fogarty H, Gaul A, Syed S, Aleksejenko N, Geoghegan R, Conroy H, et al. Adherence to hydroxyurea, health-related quality of life domains and attitudes towards a smartphone app among Irish adolescents and young adults with sickle cell disease. Ir J Med Sci 2022;191(2):809–816. doi:10.1007/s11845-021-02588-1
- Badawy SM, Thompson AA, Penedo FJ, et al. Barriers to hydroxyurea adherence and health-related quality of life in adolescents and young adults with sickle cell disease. Eur J Haematol. 2017;98(6):608–614. doi:10.1111/ejh.12878.