
ABSTRACT
Objectives
To report the hematological and molecular characteristics of a novel α-globin variant found among Chinese families.
Methods
This study was done on two unrelated families (F1 and F2). Hematological results were obtained by an automated blood cell analyzer. Hemoglobin (Hb) fraction analysis was carried out using capillary electrophoresis (CE) and high-performance liquid chromatography (HPLC). Gap-PCR and reverse dot blot (RDB) methods were used to detect the common α-thalassemia mutations in the Chinese population. The Hb variants were defined by Sanger sequencing.
Results
Hb fraction analysis of F2 cord blood using HPLC showed an abnormal peak (3.5%) of the S-window, but CE presented a 12.2% abnormal peak at zone 5(S). Similar results of CE were observed from the F1 twin's cord blood. Compared with newborns, Hb analysis of F2 father using HPLC demonstrated an abnormal peak (16.9%) of S-window and an unknown peak (0.5%) at a retention time of 4.60 min, respectively. In contrast, CE revealed a high Hb F peak at zone 7 and an unknown peak at zone 1. There was no abnormality detected with Gap-PCR and RDB in these patients. However, Sanger sequencing confirmed the presence of a new heterozygous mutation (GAC>GGC) at codon 74 of the HBA2 gene (HBA2:c.224A>G), resulting in a novel Hb variant. We named it Hb Liangqing for the birthplace of the proband.
Conclusion
This is the first report of Hb Liangqing detected by HPLC and CE. The normal hematological phenotype suggests that it may be a benign Hb variant.
Introduction
Hemoglobinopathies are the most common inherited single-gene disorders in southern China, including thalassemias and hemoglobin (Hb) variants [Citation1–3]. The molecular basis involves deletions, point mutations, and gene fusions [Citation4,Citation5]. During hematopoiesis, these alterations can form abnormal Hb variants or reduce globin chain synthesis in erythroid cells. The decrease or absence of globin chains leads to excess unpaired globin chains that precipitate in the erythrocytes, resulting in increased erythrocyte destruction or ineffective erythropoiesis [Citation6]. Due to its functional role and genetic complexity, hemoglobinopathies seriously affect human health, and patients with the disease have high morbidity and mortality [Citation7]. The clinical phenotype of hemoglobinopathies is determined by genotype. The phenotype can range from asymptomatic carrier status to mild, moderate, or severe anemia, which may be fatal [Citation8]. Therefore, accurate diagnosis of hemoglobinopathies is very important in these high-incidence regions. In many provinces of southern China, it has become a government policy to carry out hemoglobinopathy screening before marriage, pregnancy, and childbirth.
In recent years, novel Hb variants and thalassemia have been identified and described in China [Citation9–11]. In this study, we also report four cases of a novel Hb variant from two unrelated families detected during routine screening for hemoglobinopathies.
Materials and methods
Patients
This study was done on two unrelated families (F1 and F2). The proband was a newborn baby of F1 from Liangqing District, Nanning, Guangxi Zhuang Autonomous Region, China. The pregnant woman gave birth to twins in the People's Hospital of Guangxi Zhuang Autonomous Region, and the proband was the sister. They were screened for neonatal thalassemia after delivery. Another proband (male) was from the F2 who visited the Second Maternal and Child Health Care Hospital of Nanning for a routine pregnancy examination with his wife. According to the Guangxi policy requirements, they conducted a free screening test for thalassemia. The samples from two families were collected after informed consent was obtained according to the Declaration of Helsinki.
Hematological and hemoglobin fraction analysis
Blood samples from the families were collected into EDTA anti-coagulated tubes. Hematological parameters were analyzed on an automated cell counter (Sysmex XT 1800i; Sysmex Corporation, Kobe, Japan). Hemoglobin fraction analysis was done using capillary electrophoresis (CE) (Capillarys 2 Flex Piercing; Sebia, Lisses, Paris, France). Compared with CE, high-performance liquid chromatography (HPLC) was performed with the β-thalassemia Short Program (VARIANT ⅡTM; Bio-Rad, Hercules, CA, USA).
Routine genetic analysis and DNA sequencing
Gap-polymerase chain reaction (Gap-PCR) was used to detect four common deletions of α-thalassemia (including --SEA, --THAI, -α3.7, -α4.2) in the Chinese population (Yishengtang Biosciences, Shenzhen, Guangdong, China). The common three non-deletions of α-thalassemia [Hb Constant Spring (Hb CS, HBA2:c.427T>C), Hb Westmead (Hb WS, HBA2: c.369C>G), and Hb Quong Sze (Hb QS, HBA2: c.377T>C)] were carried out by PCR and reverse dot blot hybridization (PCR-RDB) kit (Yaneng Biosciences, Shenzhen, Guangdong, China). These two tests were performed according to the kit instructions. DNA sequencing identified the mutations in the HBA and HBB genes using a 3500 XL genetic analyzer sequencer (Applied Biosystems, Foster City, CA, USA). The primers and conditions for sequencing amplification were described in the previous article [Citation12,Citation13].
Results
Hematological parameters of the proband and his young sister in F1 showed no anemia, and other parameters of red blood cells were normal. In F2, the hematological parameters of the proband (male) were normal. His wife's hematological parameters were also normal, and we did not obtain complete blood count (CBC) data on his newborn (). The reference ranges of hematological parameters for adults are as follows: hemoglobin (Hb) reference 12.0–16.0 g/L, mean corpuscular volume (MCV) reference 80.0–99.0 fL, and mean corpuscular hemoglobin (MCH) reference 27.0–35.0 pg, and for newborns as follows: Hb 17.0–20.0 g/L, MCV 82.0–100.0 fL, MCH 27.0–34.0 pg.
Table 1. Hematological and Hb analysis results of patients with Hb Liangqing.
The CE results of the newborn twins presented a Hb variant peak in the cord blood of F1 (). Regretfully, these two samples could not be tested because our laboratory was not equipped with HPLC. The electrophoresis of their mother was normal, and we presume that this Hb variant was inherited from the father. Unfortunately, we did not collect the sample from the father of newborns in F1. In F2, the CE of male showed an elevated peak in the F-zone (19.0%) and a Hb variant peak (0.6%) at zone 1 ((A)). When the Hb variant and Hb F cannot be completely separated, they are easily misidentified by the instrument as Hb F. Compared with CE, HPLC revealed an abnormal Hb variant peak (16.9%) at the retention time of 4.33 min that migrated in the Hb S window and an unknown peak (0.5%) at the retention time of 4.60 min ((B)). After several months, his baby was born, and the cord blood was collected. The Hb analysis displayed a Hb variant peak using CE, as did the F1 twins ((A)). However, his HPLC chromatogram showed a 3.5% Hb variant peak in the Hb S-window at a retention time of 4.27 min ((B)).
Figure 1. Hb analysis of the proband (A) and her sister (B) cord blood with Hb Liangqing in F1 by CE. They all presented a suspicious variant peak migrating behind the Hb F peak.
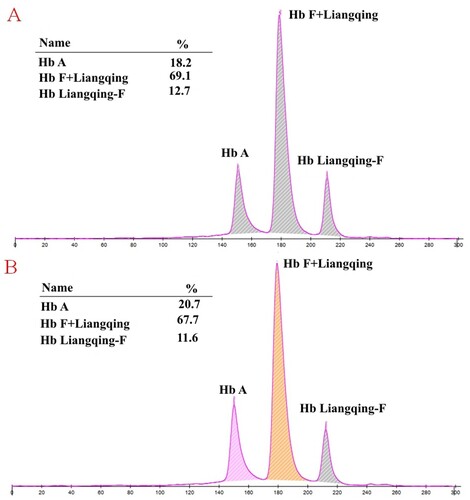
Figure 2. Hb analysis of the male in F2 with Hb Liangqing using HPLC and CE. An elevated peak in the F-zone was observed on CE (A), but HPLC revealed an abnormal peak in the S-window (B).
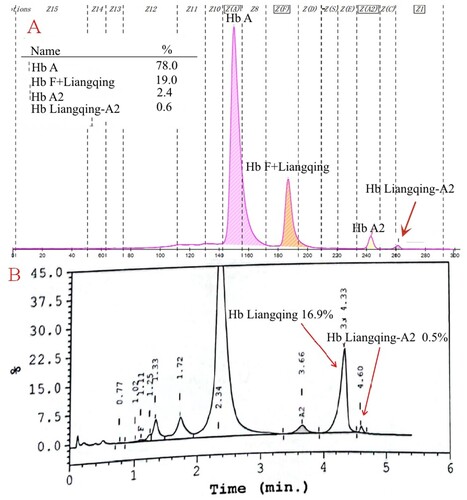
Figure 3. The Hb analysis results of the newborn in F2. CE showed a 12.2% variant peak in zone 5 (A), and HPLC displayed a low value peak (3.5%) in the S-window (B).
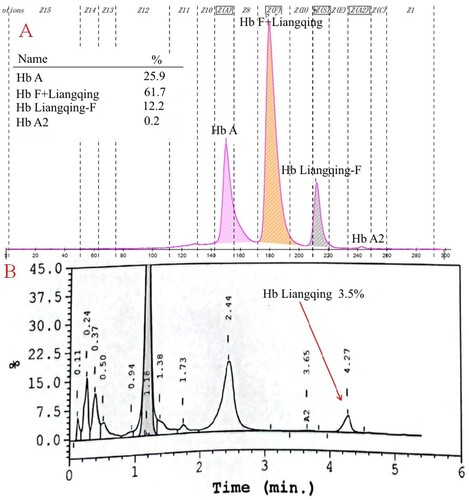
Since the CE screening results were positive, we performed routine genetic analysis to rule out the possible presence of thalassemia. We did not detect mutations in these samples by Gap-PCR and PCR-RDB. On DNA sequencing of the α-globin chain, an A>G transition was observed at codon 74 of the HBA2 gene, resulting in the replacement of the aspartic acid by a glycine residue [α74(EF3) Asp→Gly, HBA2:c.224A>G] ().
Figure 4. DNA sequencing of the proband (A) of F1, her sister (B) of F1, male of F2 (C), and infant of F2 (D). They revealed a heterozygous GAC>GGC mutation at codon 74 of the α-globin gene (arrow).
Discussion
To date, more than 1800 hemoglobinopathies have been described in two databases, HbVar (https://globin.bx.psu.edu/cgi-bin/hbvar/counter) and IthaGenes (https://www.ithanet.eu/), with more than 1300 Hb variants. Most of these Hb variants have no hematological or clinical phenotypic changes and are usually undetectable using CBC. They are only accidentally detected when performing Hb fraction analysis. In the present study, we noticed this novel Hb variant in neonatal cord blood and adult venous blood by the CE method.
In , CE presented a low value of the Hb variant in the twin's cord blood (F1). In order to investigate the location of the Hb variant, we tried to collect samples from their parents. Finally, we did not obtain the Hb variant electrophoretogram of adult blood. The HBA and HBB genes of the twins were DNA sequenced, and the same mutation was demonstrated as a substitution of GAC>GGC at codon74 of the HBA2 gene (HBA2:c.224A>G). After consulting the HbVar and Ithanet databases, this is the first mutation described at this position. We named this variant Hb Liangqing for the district of family origin of the proband. Subsequently, we uploaded the Hb Liangqing data and successfully obtained the registration from the Ithanet databases (No.3749)
Until the second family (F2), we finally collected an adult blood sample (male) with Hb Liangqing. As shown in (A), there was an additional peak (19.0%) for Hb F on CE, indicating the presence of the Hb variant. In contrast, HPLC revealed an Hb variant peak in the Hb S-window and an unknown peak, accounting for 16.9% and 0.5% of total Hb, respectively. It has been suggested that Hb Liangqing and Hb F cannot be separated using CE but must be separated using HPLC. Assume the electrophoretogram did not reveal a variant relative to Hb A2 (a clear feature of the α-globin chain mutation). In that case, it might be suspected of being hereditary persistence of fetal hemoglobin (HPFH). Of course, there is another possibility that the β-globin chain variant electrophoreses to the Hb F position and is similar to the Hb Jiangnan we reported previously [Citation14]. If this is the case, it could mislead the direction of our gene identification.
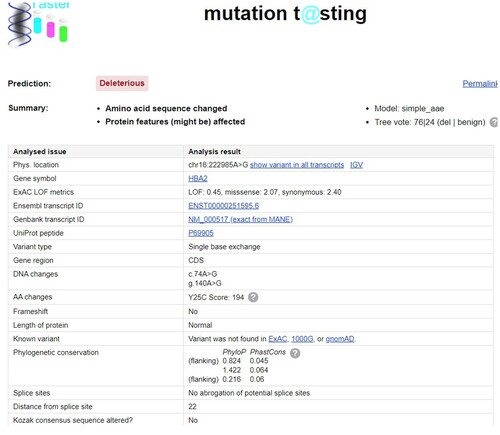
In the HBA genes (HBA1 and HBA2), six variants have been described at this location: Hb G-Pest [α74(EF3) Asp→Asn, HBA2:c.223G>A (or HBA1)], Hb Uttoxeter [α74(EF3) Asp→Tyr, HBA2:c.223G>T], Hb Q-Thailand [α74(EF3) Asp→His, HBA1:c.223G>C], Hb Les Lilas [α74(EF3) Asp→Val, HBA1:c.224A>T], Hb Lille [α74(EF3) Asp→Ala, HBA2:c.224A>C (or HBA1)], and Hb Chapel Hill [α74(EF3) Asp→Gly, HBA1:c.224A>G]. All of them have been reported to be normal structural variants or silent mutations [Citation15–17]. They can all be detected by electrophoretic and chromatographic methods. Hb G-Pest and Hb Lille migrate to the position of the S-window on HPLC, but few electropherograms of these Hb variants were described using CE. Although the mutation generates the same protein at the CD74 position in the HBA gene, Hb Chapel Hill is on the HBA1 gene, and Hb Liangqing is on the HBA2 gene. The hematological parameters of Hb Liangqing were normal, suggesting that it is also clinically benign. However, pathogenicity prediction programs (Mutation Taster) showed that Hb Liangqing might be deleterious due to the amino acid sequence change or protein features (might be) affected (). Of course, to obtain a more accurate prediction, it is necessary to combine the results of several prediction programs analyses.
Figure 5. We predicted the effect of amino acid mutation on protein. It demonstrated that Hb Liangqing might be deleterious due to the amino acid sequence change or protein features (might be) affected.
Because the screening results of her wife in the F2 were negative, the doctor did not give them any advice on prenatal diagnosis and said they only needed to follow the pregnancy examination process. Several months later, their baby was born, and cord blood was collected. After CE detection, it was suggested that the Hb variant of his father was inherited. At the same time, we also obtained the chromatogram of Hb Liangqing from the cord blood. HPLC displayed a variant peak at the S-window, with a value of 3.5% lower than that of his father (adult venous blood). We lack the hematological parameters of this newborn.
In brief, we first reported Hb Liangqing and described it as a benign variant. In addition, our study emphasizes the importance of routine evaluation of the chromatogram and electropherogram, as well as the potential analytical interference associated with Hb Liangqing. There was a clear distinction between different methods for detecting this Hb Liangqing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data in the study are shown in the figures and tables.
Additional information
Funding
References
- Zhuang J, Jiang Y, Wang Y, et al. Molecular analysis of α-thalassemia and β-thalassemia in Quanzhou region Southeast China. J Clin Pathol. 2020;73:278–282.
- Zhou B, Wang Y, Xu S, et al. Molecular spectrum of α- and β-thalassemia among young individuals of marriageable age in Guangdong Province, China. Biomed Environ Sci. 2021;34:824–829.
- He S, Qin Q, Yi S, et al. Prevalence and genetic analysis of α- and β-thalassemia in Baise region, a multi-ethnic region in southern China. Gene. 2017;619:71–75.
- Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018;70:43–53.
- Muncie HL Jr, Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009;80:339–344.
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391:155–167.
- Tanhehco YC. Gene therapy for hemoglobinopathies. Transfus Apher Sci. 2021;60:103061.
- Li Y, Liang L, Qin T, et al. Detection of hemoglobin H disease by long molecule sequencing. J Clin Lab Anal. 2022;36:e24687.
- Xu R, Li H, Yi S, et al. Identification of a novel 10.3 kb deletion causing α0-thalassemia by third-generation sequencing: pedigree analysis and genetic diagnosis. Clin Biochem. 2023;113:64–69.
- Xu A, Chen W, Xie W, et al. A novel α-globin chain variant, Hb Nanchang [HBA2: c.46G>A, Codon 15 (GGT>AGT) (Gly→Ser)], detected by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Hemoglobin. 2021;45:250–253.
- Tan X, Liu Y, Shang X, et al. A novel hemoglobin variant Hb Liaobu [α107 (G14)Val→Leu, HBA2: c.322G>C] detected by matrix-assisted laser desorption ionization-time-of-flight mass spectrometry. Hemoglobin. 2021;45:341–344.
- Li Y, Huang H, Chen Z, et al. Comparison of capillary electrophoresis and high performance liquid chromatography for detection and quantification of hemoglobin New York. Clin Chem Lab Med. 2016;54:91–95.
- Li Y, Liang L, Tian M, et al. Detection of Hb H disease caused by a novel mutation and --SEA deletion using capillary electrophoresis. J Clin Lab Anal. 2019;33:e22949.
- Liang L, Ning S, Lu X, et al. A novel mutation Hb jiangnan[β3(NA3) Leu→Lys, HBB:c.10-_11delinsAA] causing elevated Hb A2 level. Hematology. 2022;27:772–777.
- Nakatsuji T, Webber BB, Johnson SE, et al. Two rare alpha chain variants, Hb Dunn or alpha 26(A4) Asp replaced by Asn beta 2 and Hb G-Pest or alpha 274 (EF3) Asp replaced by Asn beta 2, observed in an Indian and a black newborn. Hemoglobin. 1983;7:597–599.
- Djoumessi S, Rousseaux J, Descamps J, et al. Hemoglobin Lille, alpha 2 [74(EF3) Asp replaced by Ala] beta 2. Hemoglobin. 1981;5:475–479.
- Tang B, Wang J, Qin D, et al. Hb Chapel Hill or Alpha2 74(EF3) Asp>Gly, a mildly unstable variant found in a Chinese family. Hematology. 2023;28:2187154.