
ABSTRACT
Background
Acute megakaryoblastic leukemia (AMKL) without Down syndrome (non-DS-AMKL) usually a worse outcome than DS-AMKL. Acquired trisomy 21(+21) was one of the most common cytogenetic abnormalities in non-DS-AMKL. Knowledge of the difference in the clinical characteristics and prognosis between non-DS-AMKL with +21 and those without +21 is limited.
Objective
Verify the clinical characteristics and prognosis of non-DS-AMKL with +21.
Method
We retrospectively analyzed 33 non-DS-AMKL pediatric patients and 118 other types of AML, along with their clinical manifestations, laboratory data, and treatment response.
Results
Compared with AMKL without +21, AMKL with +21 has a lower platelet count (44.04 ± 5.01G/L) at onset (P > 0.05). Differences in remission rates between AMKL and other types of AML were not significant. Acquired trisomy 8 in AMKL was negatively correlated with the long-term OS rate (P < 0.05), while +21 may not be an impact factor. Compared with the other types of AML, AMKL has a younger onset age (P < 0.05), with a mean of 22.27 months. Anemia, hemorrhage, lymph node enlargement, lower white blood cell, and complex karyotype were more common in AMKL (P < 0.05). AMKL has a longer time interval between onset to diagnosis (53.61 ± 71.15 days) (P < 0.05), and patients with a diagnosis delay ≥3 months always presented as thrombocytopenia or pancytopenia initially.
Conclusions
Due to high heterogeneity, high misdiagnosis rate, and myelofibrosis, parts of AMKL may take a long time to be diagnosed, requiring repeated bone marrow punctures. Complex karyotype was common in AMKL. +21 may not be a promising indicator of a poor prognosis.
Introduction
Acute megakaryoblastic leukemia (AMKL) is a rare subtype of acute myeloid leukemia (AML) characterized by abnormal megakaryoblasts expressing platelet-specific surface glycoprotein. AMKL is biologically heterogeneous leukemia with a poor overall prognosis [Citation1]. AMKL is extremely rare in adults, occurring in only 1% of adult AML patients. In contrast, AMKL occurs in between 4% and 15% of pediatric AML patients [Citation2]. The disease is generally divided into two major subgroups: AMKL in patients with Down syndrome (DS-AMKL) and AMKL in patients without DS (non–DS-AMKL).
AMKL is also characterized by various chromosomal abnormalities that are frequently associated with complex karyotypes and acquired clonal chromosome aberrations. DS-AMKL presents a better response to chemotherapy with a long-term survival rate of 80%. Conversely, the 5-year survival rate of non-DS-AMKL was only 10%. Researchers have been devoted to exploring the pathogenesis behind the huge difference in prognosis between these two subtypes of AMKL. Gene dosage of hematopoiesis regulatory genes on chromosome 21 has been the presumed mechanism of leukemogenesis and is related to the high chemosensitivity of DS-AMKL [Citation3–5]. In addition, acquired trisomy 21(+21) is a common chromosomal aberration in non-DS-AMKL. Though previous studies have suggested that +21 might be an indicator of poor prognosis [Citation6], many works in recent years demonstrated that a gain of chromosome 21 was not supposed to be an independent risk factor. These revised results may be possibly related to the administration of intensified chemotherapy [Citation7,Citation8].
There were controversy and limited knowledge about the clinical characteristics and prognosis of non-DS-AMKL with +21 [Citation9]. To verify the prognosis of this particular subtype of AMKL, we reported the cases of 33 patients with non-DS-AMKL, among whom ten patients were detected with +21.
Methods
Subjects
This study was approved by the Ethics Committee of the Union Hospital of Tongji Medical College, Huazhong University of Science and Technology(NO.2023-0201). In total, we defined 118 pediatric AML (expect AMKL) and 33 pediatric AMKL patients (33 non-DS-AMKL and none of DS-AMKL) from January 2012 to June 2020. All patients were diagnosed with AML referring to the 2008 or 2016 revision of the World Health Organization classification. According to the available karyotype results (n = 23), AMKL patients were divided into two groups: AMKL with +21 (n = 10) and AMKL without +21 (n = 13). Regarding differences between AMKL with +21 and AMKL without +21, we retrospectively analyzed age at onset, the time interval between onset to diagnosis, gender, clinical characteristics, signs, complete blood counts at onset, bone marrow cytogenetics findings, and treatment response.
Immunophenotyping
A four-color fluorescent direct immunoassay was used to detect the expression of surface antigens. More than 20% of antigen-expressing cells in immature myeloid cells were considered positive.
Molecular analysis
Gene detection on 3 ml of bone marrow specimens in EDTA was performed by real-time polymerase chain reaction.
Cytogenetics analysis
The karyotypes were analyzed by the G banding technique and described according to the ISCN 2009 standard.
Diagnostic criteria
The diagnostic confirmation of AMKL was determined by morphology, immunophenotyping, and cytogenetics. The morphological diagnosis was defined by the presence of ≥20% blasts in bone marrow, among which 50% were megakaryoblasts. The blasts usually show granular structure staining for PAS and negative staining for MPO. In addition, CD41 staining of bone marrow and peripheral blood may help with diagnosis. As a result of myelofibrosis, dry tap aspiration or dilution of bone marrow is common in AMKL. Hence, flow immunotyping often fails to work. The blasts of AMKL usually express markers CD41, CD42, and CD61. The aberrations of gain of chromosome 8, gain of chromosome 21, monosomy 7, t (1;22), and t (11;12) are common in AMKL.
Treatment protocol
All patients received routine chemotherapy for AML. The 1st induction phase contained DAE (Daunorubicin (DNR), Cytarabine (Ara-C), and Etoposide) or DAH (DNR, Ara-C, Homoharringtonine (HHT)) randomly. The 2nd induction phase contained IAE (Idarubicin (IDA), Ara-C, and Etoposide) or IAH (IDA, Ara-C, HHT). The consolidation phase includes MA (Mitoxantrone (MIT), high-dose Ara-C)-HA (HHT, Ara-C)-CLASP (Ara-C, L-asparaginase(L-ASP)), and the chemotherapy dose was adjusted according to the risk level. During the maintenance phase, patients were randomized to different treatment groups of Ara-C with 6-mereaptopurine (6-MP) or all-trans retinoic acid (ATRA) with 6-MP. Some patients with or without CR were offered hematopoietic Stem Cell Transplantation (HSCT).
Response evaluation
Evaluations of bone marrow aspirates were performed after the 1st and 2nd inductions. The evaluation was conducted after each course of consolidation. The evaluation was conducted every six months during maintenance treatment until the end of chemotherapy. CR was defined as bone marrow with <5% blasts and evidence of the regeneration of normal hematopoietic cells; and minimal residual disease (MRD) ≤10−4. Relapse was defined as the presence of ≥5% blasts in the bone marrow or extramedullary relapse. The last follow-up date was 10 December 2021, follow-up time range from one to 104 months.
Statistical analysis
Statistical analyses were carried out using SPSS26. Measurement data were analyzed using the independent sample t-test or Wilcoxon rank-sum test. The prognostic factors associated with AMKL were analyzed by binary Logistic regression. Overall survival (OS) probability was defined as the time from diagnosis to death by any cause. The survival distribution between groups was performed using the Kaplan-Meier method and was compared using the log-rank test. P values less than 0.05 were considered statistically significant.
Results
Clinical characteristics
From January 2012 to June 2020, 33 patients (including ten patients with +21 and 13 patients without +21) were diagnosed with AMKL from four centers. Interestingly, no one was diagnosed with DS-AMKL. There were more females than males, with a male-to-female ratio of 1:1.54. The median onset age of AMKL in our patients was 17 months (IQR, 12–29 months). 87.8(29/33) of the patients in our study presented thrombocytopenia initially. Bone marrow aspiration was difficult in 30.3%(10/33) patients. Compared with the other types of AML during this time interval, AMKL has a younger onset age (P < 0.05), with a mean of 22.27 months. Anemia, hemorrhage, less lymph node enlargement, and lower white blood count were more common in AMKL at onset (P < 0.05). The time interval between onset to diagnosis in AMKL was significantly longer than in AML (P < 0.05). The clinical manifestations and laboratory data of the patients are shown in .
Table 1. Comparison of AMKL versus other AMLs.
Compared to AMKL without +21, we observed that AMKL with +21 had a lower platelet count at onset, but the difference was not significant (P = 0.594). Age, gender, the time interval between onset to diagnosis, blood count, and response to therapy between the two groups had no significant difference ().
Table 2. Comparison of AMKL with acquired trisomy 21 versus AMKL without acquired trisomy 21.
Immunophenotyping
Results of bone marrow flow cytometry immunotyping in 25 of 33 AMKL patients were available for analysis. 91.3%(21/23) of patients were positive for CD41a and CD61, and 95.7%(22/23) were positive for CD42b. In addition, CD33 (88%(22/25)), CD34 (78.3%(18/23)), CD36 (73.3%(11/15)), CD117 (86.4%(19/22)), CD13 (78.3%(18/22)), HLA-DR (36.4%(8/23)), CD3 (31.3%(5/16)), CD56 (68.2%(15/22)), and CD7 (34.8%(8/23)) were detected. The difference in CD36 positive rates between AMKL with +21 and AMKL without +21 was not significant.
Molecular analysis
Gene detection was performed in 27 AMKL patients, among which two cases were detected with elevated EVI1 expression, three cases with elevated WT1 expression, three cases with both elevated EVI1 and WT1 expression, one case carried MPL mutation and high EVI1 and WT1 expression, one case carried GATA1 mutation and high EVI1, WT1 expression, and one case carried MLL/AF9 and high EVI1 expression ().
Table 3. Cytogenetic analysis, molecular analysis results of 33 AMKL cases.
Cytogenetics analysis
Karyotype analysis was performed in 23 of the AMKL patients. Among the 23 evaluable patients, normal karyotypes (n = 6, 26.1%) and complex karyotypes (n = 13, 56.5%) were observed, ten patients (43.5%) had acquired +21 aberrations (). Compared with other types of AML, complex karyotype was more common in AMKL (P < 0.05).
Response to therapy
Among the 33 patients with AMKL, 12 patients gave up treatment, one patient died before therapy. 20 patients received therapy, among which two patients only received blood transfusions and anti-infection therapy for their reasons. Six patients (35.3%) achieved CR after 1st induction, and eight patients (53.3%) achieved CR after 2nd induction. Seven patients received HSCT. Eleven patients survived, among which six patients lived without HSCT (). As of the follow-up time, the median survival time of the 20 patients who received treatment was 17.5 months (range frome1 to 104 months).
Figure 1. Outcomes of 18 patients. AMKL, acute megakaryocytic leukemia; HSCT, hematopoietic stem cell transplantation; CR, complete remission.
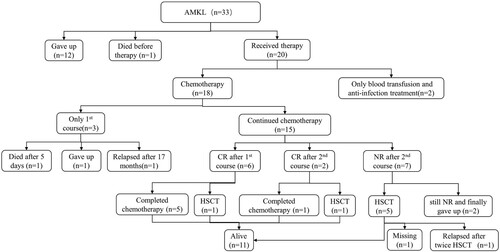
The difference in remission rate between AMKL and other types of AML, and the difference in remission rate between AMKL with +21 and AMKL without +21, were not significant ( and ).
Impact factors on remission rate after 1st and 2nd induction were analyzed by logistic regression analysis. Factors (including age, gender, time internal ≥30 days, extramedullary infiltration, WBC ≥ 10G/L, PLT, CD36 positive, WT1, complex karyotype, +21, +19, and +6) had no significant effect on remission rate of 1 and 2 courses. We found that EVI1 was negatively correlated with the remission rate after 2nd induction, but not significant (P = 0.085).
Long-term survival
Factors (including gender, age, the time interval between onset to diagnosis, extramedullary infiltration, white blood cell count at diagnosis, CD36, WT1 expression, complex karyotypes, +6, and +19), did not affect survival. patients with +21 (P log-rank = 0.185), and patients with no remission after 1st-course therapy (P log-rank = 0.053), tend to have a lower OS rate, patients with HSCT tend to have a better OS rate than patients without HSCT (P log-rank = 0.102), but the data were not statistically significant. However, of 23 patients with available cytogenetics results, patients with +8 had worse OS rates compared with patients without +8 (P log-rank = 0.005). Moreover, patients with no remission after 2nd-course therapy also had a worse OS rate than patients who achieved CR after 2nd course (P log-rank = 0.028) ( and ).
Discussion
AMKL is a subtype of AML with high heterogeneity. In this study, the male-to-female ratio of 1:1.54 was different from previous studies that mentioned that AMKL was more common in males [Citation10]. The mean onset age of AMKL was 22.27 months, which was significantly lower than other types of AML. Besides, chromosomal abnormalities were found to be frequent in AMKL, most of which are accompanied by complex karyotypes. The clinical features of the patients in our study were similar to other studies [Citation10,Citation11]. Interestingly, there was no significant difference in early response rates between AMKL and other types of AML patients in this study.
The time interval from onset to diagnosis of AMKL was significantly longer than that of other types of AML patients (P < 0.05). To further investigate this, the medical histories of patients with a long-term interval from onset to diagnosis were reviewed. Patients with a diagnosis interval of more than three months always presented with thrombocytopenia or pancytopenia at initial, moreover, they were commonly misdiagnosed with immune thrombocytopenia or aplastic anemia, which result in inappropriate treatment and prolonged delay in correct diagnosis. Ultimately, the diagnosis was confirmed through repeated bone marrow puncture, with one patient undergoing five bone punctures before receiving a confirmed diagnosis. Of the 33 AMKL patients, 10 presented with bone puncture dry aspiration, six of whom underwent bone marrow biopsy. Four out of the six patients who underwent biopsy presented with myelofibrosis (MF). Although not statistically significant, the time interval from onset to diagnosis was longer in these patients with MF (mean 93.5 ± 58.5 days) than other AMKL patients. A prolonged time interval from onset to diagnosis and the presence of MF may lead to delayed treatment and its associated poor outcomes [Citation12].
Megakaryoblasts in AMKL are positive for markers of megakaryoblastic lineages such as CD41a or CD61 in flow cytometry. 11 patients in this study were detected CD36 positive, and five of them achieved favorable treatment outcomes. Studies had demonstrated that DS-AMKL cases showed high expression of CD36 and were very sensitive to cytarabine and daunorubicin. Both DS-AMKL and good prognosis non-DS-AMKL patients demonstrate high expression of CD36 [Citation13,Citation14].
AMKL is characterized by a range of chromosomal abnormalities, with specific chromosomal abnormalities and gene mutations that could be valuable for stratification. Children with DS are >100 times more likely to develop AMKL. The altered dosage of genes associated with hematopoiesis, megakaryopoiesis, and leukemia on chromosome 21 may contribute to aberrant blood development. Acquisition of additional copies of chromosome 21 is the most common chromosomal abnormality in non-DS-AMKL [Citation15,Citation16]. The mean of platelet counts in AMKL patients with +21 was lower than AMKL without +21 in our study, which has not been previously reported. Thrombocytopenia was non-randomly found in individuals with deletion 21 syndrome, which may be related to genes related to platelet generation on chromosome 21 [Citation17].
Although DS patients were not excluded in our study, the incidence (0%) of DS-AMKL in our study is significantly lower than other studies (range from 1% ∼53%) [Citation18]. Trisomy 21 with GATA1 mutation is common in transient myeloproliferative disorder and is related to DS-AMKL [Citation19–22]. In our study, only one patient was diagnosed with DS-AML(M2) with GATA1 mutation. Unfortunately, he did not pursue treatment after his diagnosis. Interestingly, GATA1 mutation had also been found in non-DS-AMKL with acquired trisomy 21 [Citation9,Citation23–25]. We did observe that one patient with AMKL who had acquired trisomy 21 and GATA1 mutation achieved CR after 2nd induction. Subsequently, she underwent allogeneic HSCT and was in an event-free survival state after a 25-month follow-up. Intriguingly, in a study by Jana Schweitze, all six patients with non-DS-AMKL and GATA1 mutations were in continuous remission after treatment, suggesting a similar biology and treatment response of AMKL with acquired or constitutional trisomy 21 in conjunction with GATA1 mutations, which may indicate a relatively favorable prognosis [Citation8]. A recent study showed that GATA1 mutations can be detected in 11% of non-DS-AMKL patients and that low-intensity chemotherapy may also be effective in these patients [Citation15,Citation26]. More evidence is needed to demonstrate the relation between GATA1 mutation and non-DS-AMKL.
The EVI1 and WT1 genes have been found to be widely over-expressed in most cases of AML. In this study, 25.9% (7/27) of AMKL patients had high EVI1 expression, and 29.6% (8/27) had high WT1 expression. However, the prognosis of these patients was not significantly different from others. The WT1 gene product is known to exhibit both oncogenic and tumor suppressor properties. Although several studies initially showed an association between elevated WT1 expression and low survival in adult AML, recent studies on diagnostic WT1 expression had revealed no significant prognostic impact [Citation27]. The EVI1 gene, located in chromosomal band 3q26, is a transcription factor with stem cell-specific expression pattern and is essential for the regulation of self-renewal of hematopoietic stem cells. High expression of EVI1 can inhibit the response of granulocytic cells to colony-stimulating factors, as well as inhibit the transcription induced by the GATA-1 transcription factor, and hinder myeloid cell maturation and differentiation. Current studies suggest that high expression of EVI1 is associated with poor prognosis in AML, which has not been observed in AMKL patients [Citation28,Citation29].
56.5% of non-DS-AMKL patients in our study were diagnosed with complex cytogenetic aberrations, including +21, +19, +8, and +6. F Stölzel analyzed 3526 AML patients and found that patients with complex karyotypes had a risk of reduced overall survival compared with patients with a normal karyotype [Citation30]. In our study, AMKL patients with complex karyotype and those with + 21 had a relatively lower overall long-term survival (). However, +8 was confirmed to be associated with low survival rate (), the lower survival rate of patients with + 21 in our study possibly due to the fact that 50% of patients with + 21 also had +8. To eliminate the potential effect of +8, we compared the survival rates of patients with +21 and +8 and patients with +21 but without +8. It was found that patients with +21 but without +8 had better outcomes ( and ). Overall, no significant difference of survival rates between non-DS-AMKL with +21 and non-DS-AMKL without +21 was found in our study, indicating that +21 may have no significant impact on the prognosis of non-DS-AMKL. Patients who achieved CR after 2nd induction and those who underwent HSCT had higher long-term survival rates in our study, consistent with current research [Citation31,Citation32]. Therefore, achieving CR in the early stages of treatment, and undergoing HSCT could improve the prognosis of non-DS-AMKL.
Figure 2. Kaplan-Meier Estimates of OS in the 33 AMKL patients. A: Patients without +21 VS patients with +21. B: Patients without +8 VS patients with +8. C: Patients without complex karyotype VS patients with complex karyotype. D: Patients achieved CR after 2nd course VS patients didn’t achieve CR after 2nd course. E: Patients received chemotherapy VS patients received HSCT.
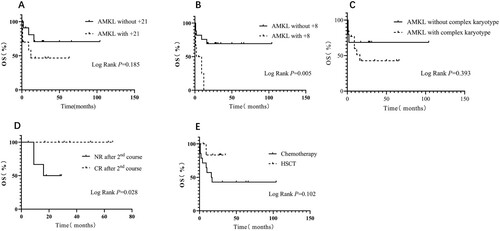
Figure 3. Kaplan-Meier Estimates of OS in the 33 AMKL patients. A: Patients with +21 but without +8 VS patients with +21 and +8. B: Patients with +21 but without +8 VS other patients. C: Patients with +21 only VS other patients.

Our study had several limitations. Although our longest follow-up time was 104 months, the time for observation in our study was still not long enough. Half of the patients lived in disease-free conditions without HSCT in our study, which is different from previous studies. Additionally, the incidence of AMKL is very low and the number of patients is relatively limited in our work. Therefore, a longer follow-up period and larger sample sizes are needed to obtain a clearer understanding of the patients’ prognosis, verify the role of chromosome 21 in AMKL, and improve clinicians’ understanding of AMKL.
Conclusion
Non-DS-AMKL is a disease with a poor prognosis. With the improvements in diagnosis and intense chemotherapy, the survival of patients with AMKL has been greatly improved. Chromosome 21 plays an important role in the occurrence and development of hematologic malignancies, especially in AMKL. In conclusion, acquired chromosome 21 aberrations may not be an indicator of poor prognosis. Therefore, cytogenetic analysis is helpful for AMKL risk stratification, prognostic judgment, and individualized treatment.
Author contributions
The study was originally and conceptually developed and supervised until its completion by XW, WZ performed the data collection, and data analyses and wrote the manuscript; JD performed the data collection, XW, HC, FZ, and YQ helped perform the analysis with constructive discussions. HL, JL, HY, JX, JH and QH provided data on patients. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Acknowledgments
We thank all the doctors in Department of Pediatrics at Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, and Department of Pediatrics at Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and Department of Hematology at Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, and Department of Pediatrics at The Central Hospital of Enshi Autonomous Prefectureand, and all the patients included in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi:10.1182/blood-2009-03-209262
- Gruber TA, Downing JR. The biology of pediatric acute megakaryoblastic leukemia. Blood. 2015;126(8):943–949. doi:10.1182/blood-2015-05-567859
- Athale UH, Razzouk BI, Raimondi SC, et al. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution’s experience. Blood. 2001;97(12):3727–3732. doi:10.1182/blood.V97.12.3727
- Garnett C, Cruz Hernandez D, Vyas P. GATA1 and cooperating mutations in myeloid leukaemia of Down syndrome. IUBMB Life. 2020;72(1):119–130. doi:10.1002/iub.2197
- Roberts I, O’Connor D, Roy A, et al. The impact of trisomy 21 on foetal haematopoiesis. Blood Cells Mol Dis. 2013;51(4):277–281. doi:10.1016/j.bcmd.2013.07.008
- Wan TS, Au WY, Chan JC, et al. Trisomy 21 as the sole acquired karyotypic abnormality in acute myeloid leukemia and myelodysplastic syndrome. Leuk Res. 1999;23(11):1079–1083. doi:10.1016/S0145-2126(99)00117-4
- Schweitzer J, Zimmermann M, Rasche M, et al. Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol. 2015;94(8):1327–1336. doi:10.1007/s00277-015-2383-2
- Inaba H, Zhou Y, Abla O, et al. Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood. 2015;126(13):1575–1584. doi:10.1182/blood-2015-02-629204
- Ono R, Hasegawa D, Hirabayashi S, et al. Acute megakaryoblastic leukemia with acquired trisomy 21 and GATA1 mutations in phenotypically normal children. Eur J Pediatr. 2015;174(4):525–531. doi:10.1007/s00431-014-2430-3
- Marques-Piubelli ML. Acute megakaryoblastic leukemia with t(1;22)(p13.3;q13.1); RBM15-MKL1 mimicking hepatoblastoma in an infant: The role of karyotype in differential diagnosis. Pediatr Blood Cancer. 2020 Mar;67(3):e28111. doi: 10.1002/pbc.28111.
- Maarouf N, Mahmoud S, Khedr R, et al. Outcome of childhood acute megakaryoblastic leukemia: Children’s Cancer Hospital Egypt 57357 experience. Clin Lymphoma Myeloma Leuk. 2019;19(3):e142–e152. doi:10.1016/j.clml.2018.12.011
- Zhao G, Wu W, Wang X, et al. Clinical diagnosis of adult patients with acute megakaryocytic leukemia. Oncol Lett. 2018;16(6):6988–6997. doi: 10.3892/ol.2018.9501
- Khan I, Malinge S, Crispino J. Myeloid leukemia in Down syndrome. Crit Rev Oncog. 2011;16(1-2):25–36. doi:10.1615/CritRevOncog.v16.i1-2.40
- Savasan S, Buck S, Raimondi SC, et al. CD36 (thrombospondin receptor) expression in childhood acute megakaryoblastic leukemia: in vitro drug sensitivity and outcome. Leuk Lymphoma. 2006;47(10):2076–2083. doi:10.1080/10428190600773180
- McNulty M, Crispino JD. Acute megakaryocytic leukemia. Cold Spring Harb Perspect Med. 2020;10(2):a034884. doi:10.1101/cshperspect.a034884
- Evans EJ Jr, DeGregori J. Dissecting stepwise mutational impairment of megakaryopoiesis in a model of Down syndrome-associated leukemia. J Clin Invest. 2022;132(14). doi:10.1172/JCI161659
- Huret JL, Léonard C. Chromosome 21 and platelets: a gene dosage effect? Clin Genet. 1997;51(2):140–141. doi:10.1111/j.1399-0004.1997.tb02442.x
- Hama A, Yagasaki H, Takahashi Y, et al. Acute megakaryoblastic leukaemia (AMKL) in children: a comparison of AMKL with and without Down syndrome. Br J Haematol. 2008;140(5):552–561. doi:10.1111/j.1365-2141.2007.06971.x
- Lee WY, Weinberg OK, Pinkus GS. GATA1 is a sensitive and specific nuclear marker for erythroid and megakaryocytic lineages. Am J Clin Pathol. 2017;147(4):420–426. doi:10.1093/ajcp/aqx018
- Crispino JD, Horwitz MS. GATA factor mutations in hematologic disease. Blood. 2017;129(15):2103–2110. doi:10.1182/blood-2016-09-687889
- Bourquin JP, Subramanian A, Langebrake C, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci U S A. 2005;103(9):3339–3344. doi:10.1073/pnas.0511150103
- Alejo-Valle O, Weigert K, Bhayadia R, et al. The megakaryocytic transcription factor ARID3A suppresses leukemia pathogenesis. Blood. 2022;139(5):651–665. doi:10.1182/blood.2021012231
- Lopez CK, Malinge S, Gaudry M, et al. Pediatric acute megakaryoblastic leukemia: multitasking fusion proteins and oncogenic cooperations. Trends Cancer. 2017;3(9):631–642. doi:10.1016/j.trecan.2017.07.003
- Wang SA, Hasserjian RP. Acute erythroleukemias, acute megakaryoblastic leukemias, and reactive mimics: A guide to a number of perplexing entities. Am J Clin Pathol. 2015;144(1):44–60. doi:10.1309/AJCPRKYAT6EZQHC7
- Shin M-G, Choi H-W, Kim H-R, et al. Tetrasomy 21 as a sole acquired abnormality without GATA1 gene mutation in pediatric acute megakaryoblastic leukemia: a case report and review of the literature. Leuk Res. 2008;32(10):1615–1619. doi:10.1016/j.leukres.2008.02.010
- Terui K, Toki T, Taga T, et al. Highly sensitive detection of GATA1 mutations in patients with myeloid leukemia associated with Down syndrome by combining Sanger and targeted next generation sequencing. Genes Chromosomes Cancer. 2020;59(3):160–167. doi:10.1002/gcc.22816
- Ho PA, Alonzo TA, Gerbing RB, et al. The prognostic effect of high diagnostic WT1 gene expression in pediatric AML depends on WT1 SNP rs16754 status: report from the Children’s Oncology Group. Pediatr Blood Cancer. 2014;61(1):81–88. doi:10.1002/pbc.24700
- De Braekeleer M, Le Bris MJ, De Braekeleer E, et al. 3q26/EVI1 rearrangements in myeloid hemopathies: a cytogenetic review. Future Oncol. 2015;11(11):1675–1686. doi:10.2217/fon.15.64
- Jo A, Mitani S, Shiba N, et al. High expression of EVI1 and MEL1 is a compelling poor prognostic marker of pediatric AML. Leukemia. 2015;29(5):1076–1083. doi:10.1038/leu.2015.5
- Stolzel F, Mohr B, Kramer M, et al. Karyotype complexity and prognosis in acute myeloid leukemia. Blood Cancer J. 2016;6:e386. doi:10.1038/bcj.2015.114
- Huang J, Hu G, Suo P, et al. Unmanipulated haploidentical hematopoietic stem cell transplantation for pediatric de novo acute megakaryoblastic leukemia without Down syndrome in China: a single-center study. Front Oncol. 2023;13:1116205. doi:10.3389/fonc.2023.1116205
- Hama A, Taga T, Tomizawa D, et al. Haematopoietic cell transplantation for children with acute megakaryoblastic leukaemia without Down syndrome. Br J Haematol. 2023 May;201(4):747–756. doi: 10.1111/bjh.18691