
ABSTRACT
Background
In China, conventional genetic testing methods can only detect common thalassemia variants. Accurate detection of rare thalassemia is crucial for clinical diagnosis, especially for children that need long-term blood transfusion. This study aims to explore the application value of third-generation sequencing (TGS) in the diagnosis of rare thalassemia in children with anemia.
Methods
We enrolled 20 children with anemia, excluding from iron deficiency anemia (IDA). TGS was employed to identify both known and novel thalassemia genotypes, while sanger sequencing was used to confirm the novel mutation detected.
Results
Among the 20 samples, we identified 5 cases of rare thalassemia. These included β−4.9 (hg38,Chr11:5226187-5231089) at HBB gene, α−91(HBA2:c.*91delT), αCD30(HBA2:c.91-93delGAG), Chinese Gγ+(Aγδβ)0(NG_000007.3: g .48795-127698 del 78904) and delta – 77(T > C)(HBD:c.-127T>C). Notably, the –SEA/α−91α genotype associated with severe non-deletional hemoglobin H disease (HbH disease) has not been previously reported. Patients with genotypes β654/β−4.9 and –SEA/α−91α necessitate long-term blood transfusions, and those with the –SEA/αCD30α, Chinese Gγ+(Aγδβ)0 and delta thalassemia demonstrate mild anemia.
Conclusions
TGS demonstrates promising potential as a diagnostic tool for suspected cases of rare thalassemia in children, especially those suspected to have transfusion-dependent thalassemia (TDT).
1. Introduction
Thalassemia is a common monogenic genetic disease caused by defects in the globin gene. The composition of globin undergoes dynamic changes during childhood [Citation1]. Accurate diagnosis of anemia based solely on hematological phenotypes is challenging, highlighting the importance of precise genetic testing. At present, the molecular diagnosis of thalassemia uses methods such as Gap-PCR and reverse dot blot (RDB) hybridization. However, these methods can only detect the common mutation types present in the Chinese population. The application of sanger sequencing and next-generation sequencing (NGS) has revealed some rare mutations [Citation2–4]. In recent years, TGS which has the advantages of long read length, single molecular resolution, and fast detection speed, has been gradually applied in the field of thalassemia detection [Citation5,Citation6].
In this study, we employed TGS to successfully identify two cases of HbH disease caused by–SEA/αCD30α and –SEA/α−91α which belongs to TDT, another case of TDT caused by β654/β−4.9, one case of heterozygote Chinese Gγ+(Aγδβ)0-thalassemia, and one case of delta - 77(T > C). Additionally, α−91 is a novel mutation at HBA2 gene. The results from our study provide reliable information for genetic counseling and thalassemia diagnosis.
2. Methods
2.1. Patients
We recruited a total of 20 subjects suspected rare thalassemia from our hospital. Among them, 17 cases presented with microcytic hypochromic anemia but were negative for routine thalassemia genetic testing. One case was suspected of having HbH disease, but routine testing showed a heterozygosity for – SEA. Two cases were suspected TDT patients who required blood transfusion, but routine genetic testing indicated mild thalassemia. All cases were excluded for IDA. This study was approved by the Ethics Review Board of Shenzhen Children’s Hospital (No.202211602).
2.2. Routine gene analysis
Genomic DNA was purified from whole blood using a DNA isolation kit and common thalassemia genes were detected using a corresponding detection kit (Yilifang, Shenzhen, People’s Republic of China).
Gap-PCR was utilized to detect the four α-thalassemia deletions (–SEA, -α3.7, -α4.2, and –THAI). PCR-RDB was used to identify the three non-deletional α-thalassemia mutations (Hb Constant Spring [Hb CS], Hb Quong Sze [Hb QS], and Hb Westmead [Hb WS]). For the molecular diagnosis of common β-thalassemia, PCR-RDB was performed to detect the nineteen β-globin gene mutations known to occur in the Chinese population, including codons 41/42 (–CTTT), IVS-II-654 (C > T), –28 (A > G), codons 71/72 (+A), codon 17 (A > T), codon 26(βE) (G > A), codon 31 (–C), codon37(TGG > TAG), codon 27/28 (+C), IVS-I-1 (G > T), codon 43 (G > T), –32 (C > A), initiation codon (ATG > AGG), –30 (T > C), codons 14/15 (–G), Cap +1 (A > C), –29 (A > G), –50(G > A), and IVS-I-5 (G > C).
2.3. TGS and Sanger sequencing
Samples were sent to an independent laboratory (Berry Genomics, Fuzhou, China), Genomic DNA samples were prepared using the DNA Blood Mini Kit from QIAGEN. After evaluating the DNA quality, these samples underwent multiplex long PCR using the assay kit from Berry Genomics. Each PCR product was purified and subjected to end repair before being ligated with a unique identifying barcoded adaptor. Subsequently, single-molecule real-time (SMRT) bell libraries were prepared using the Sequel Binding and Internal Ctrl Kit 3.0 from Pacific Biosciences. The DNA-polymerase complexes were loaded onto SMRT cells and sequenced on the PacBio Sequel platform. To obtain aligned subreads, raw reads were processed using the Lima application, followed by circular consensus sequencing (CCS) software application. The filtered CCS reads were aligned to the human reference genome (hg19) using NGMLR (version 0.2.6) or BLASR, and variants were called using Free Bayes (version 1.2.0), depending on the specific objectives. Alignments of variant and wild-type molecules were visualized using the Integrative Genomics Viewer (IGV).
Sanger sequencing was performed on an ABI PRISM 3730 XL Sequencer (Applied Biosystems, Foster City, CA).
3. Results
In a group of 20 cases, the common genotypes of 2 cases suspected of TDT were β 654/ βN and – SEA/αα, respectively. 1 case was suspected HbH disease which the H-band was seen in hemoglobin electrophoresis, as shown in , but routine testing showed – SEA/αα. 17 cases were undetected globin gene mutations. However, rare thalassemia was detected in 5 cases using TGS.
Table 1. Genotypes and phenotypes of 5 cases of rare thalassemia.
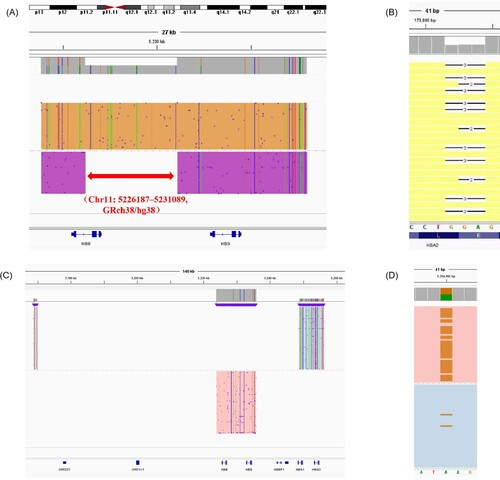
As shown in , 1 case of TDT caused by β654/β−4.9 was identified. The β−4.9 (hg38, Chr11:5226187-5231089) is at HBB gene ( (A)) .1 case of –SEA/αCD30α was identified. The αCD30(HBA2:c.91-93delGAG) is at HBA2 gene ( (B)). 1 case of heterozygote Chinese Gγ+(Aγδβ)0-thalassemia (NG 000007.3: g .48795-127698 del 78904) was identified ( (C)). 1 case of delta -thalassemia (HBD:c.−127T > C) was identified ( (D)).
Figure 1. Integrative Genomics Viewer plots identified by TGS. The red arrow represents −4.9 kb deletion at chr16: 5226187–5231089 (A). The blank space represents αCD30(-GAG) in HBA2 (B). The colored part represents all the deletion of HBB and HBD and partial deletion of HBG(C). The brown color represents delta-thalassemia (c.−127T > C) in HBD (D).
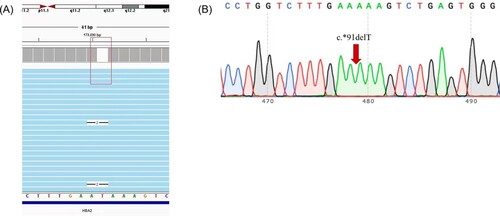
As shown in , 1 case of severe non-deletional HbH disease caused by –SEA/α−91α mutation in HBA2 gene was identified. The α-91(HBA2:c.*91delT) locates at HBA2 gene ( (A)). To our knowledge, the mutation of had never been described before. Sanger sequencing confirmed this novel mutation ( (B)).
Figure 2. The TGS result and validation of α−91. Integrative Genomics Viewer plot identified by TGS. The red box represents α−91 (-T) in HbA2(A). Sanger sequencing confirmed for the novel mutation (B).
It should be noted that 2 cases with TDT no longer have hematological indicators that can serve as reference values due to their long-term blood transfusion history. As a result, these patients’ data are not included in the table1. Although both –SEA/α−91α and –SEA/αCD30α belong to non-deletional H disease, it can be seen from that –SEA/αCD30α only leads to mild anemia. Heterozygote Chinese Gγ+(Aγδβ)0-thalassemia and delta -thalassemia (HBD:c.−127T > C) cause mild anemia, with the latter having a lower HbA2 level of 1.1%.
4. Discussion
Hematological indicators such as mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), fetal hemoglobin (HbF), and HbA2 are sensitive screening tools for thalassemia [Citation7]. However, in growing children, hemoglobin levels undergo dynamic changes, leading to reduced sensitivity of these hematological indicators. Deletional β-thalassemias like Chinese Gγ+(Aγδβ)0 and SEA-HPFH have been shown to increase HbF levels [Citation8]. However, in children where HbF has not completely disappeared, determining whether it is thalassemia based on HbF alone becomes challenging.
Clinical practice has confirmed that α-thalassemia and delta-thalassemia are the main causes of reduced HbA2 levels [Citation9]. In this study, a case with HbA2 level of 1.1% (Case 11) was investigated after excluding iron deficiency anemia and α-thalassemia, indicating the need to consider the presence of delta-thalassemia.
For the detection of Chinese Gγ+(Aγδβ)0 and delta-thalassemia, techniques like Gap-PCR or Sanger sequencing require specific primer designs and have limited detection ranges.
Single-molecule real-time sequencing (SMRT sequencing) is a third-generation sequencing technology that utilizes a specialized sequencing platform and chemical methods. This method relies on fluorescence signals emitted by DNA polymerase during the amplification process for sequencing.
In SMRT sequencing, the DNA sample is fragmented either by restriction enzymes or random fragmentation, and these fragments are attached to a transparent array surface. An enzyme called DNA polymerase, which carries a fluorescent label capable of emitting fluorescence signals, is introduced onto this surface. As the DNA polymerase replicates the DNA strands, the fluorescence signals are recorded and detected.
By continuously monitoring the changes in fluorescence signals in real-time, multiple reads can be obtained from a single DNA molecule. This approach eliminates the need for amplifying DNA fragments into large quantities, reducing the potential for introduced errors seen in traditional sequencing techniques. Additionally, since SMRT sequencing utilizes long DNA molecules for sequencing, longer read lengths can be achieved, aiding in addressing challenges associated with complex genomes or repetitive sequences.
Overall, TGS offers advantages such as long read length, high accuracy, and no GC preference [Citation10]. Additionally, TGS can determine whether mutations are present in a cis or trans configuration. The presence of two mutation points on separate alleles (trans-configuration) may lead to severe anemia. In this study, β654 and β−4.9 were located on separate alleles, causing TDT.
Children with TDT require regular transfusions for survival and include individuals with β-thalassemia major, severe HbE/β-thalassemia, severe non-deletional HbH disease, and survivors of Hb Bart’s hydrops fetalis syndrome [Citation11]. The diagnosis of TDT requires comprehensive analysis of clinical history, hematological indicators, and genotypes. However, when mild thalassemia occurs alongside other blood diseases like hereditary spherocytosis, the patient may exhibit hemolysis similar to TDT, leading to potential misdiagnosis [Citation12]. Therefore, precise genetic testing is crucial for accurate diagnosis.
In addition to the TDT case, TGS confirmed one case of severe non-deletional HbH disease (–SEA/α−91α). HbH disease is divided into deletional and non-deletional types, with the non-deletional type being more severe [Citation13,Citation14]. In this study, –SEA/α−91α corresponded to the non-deletional type of HbH disease, characterized by splenomegaly and blood transfusion dependence since infancy. However, the –SEA/αCD30α in this study only presented mild anemia, consistent with a report by Chen et al. [Citation15], but not with another report by Ma et al. [Citation16]. The heterogeneity of hemoglobin H disease may be related to race or modified genes, requiring further research.
To date, the −4.9 kb deletion in the HBB gene resulting in mild thalassemia has only been reported by Chen et al. using TGS in 2022 [Citation17]. Furthermore, the novel α−91(HBA2:c.*91delT) mutation identified in this study has not been previously reported.
In summary, TGS is a valuable choice for diagnosing children suspected of having rare thalassemias, particularly those suspected of having TDT.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149(2):181–194. doi:10.1111/j.1365-2141.2010.08105.x
- Huang H, Xu L, Chen M, et al. Molecular characterization of thalassemia and hemoglobinopathy in southeastern China. Sci Rep. 2019;9(1):3493. doi:10.1038/s41598-019-40089-5
- Zhuang J, Jiang Y, Wang Y, et al. Molecular analysis of α-thalassemia and β-thalassemia in quanzhou region southeast China. J Clin Pathol. 2020;73(5):278–282. doi:10.1136/jclinpath-2019-206179
- Chen J, Lin S, Gan J, et al. A novel β-thalassemia variant at HBB:c.14delC (codon 4, -C) identified via next-generation sequencing. Hematology. 2020;25(1):400–404. doi:10.1080/16078454.2020.1841920
- Peng C, Zhang H, Ren J, et al. Analysis of rare thalassemia genetic variants based on third-generation sequencing. Sci Rep. 2022;12(1):9907. doi:10.1038/s41598-022-14038-8
- Xu L, Mao A, Liu H, et al. Long-Molecule sequencing. J Mol Diagn. 2020;22(8):1087–1095. doi:10.1016/j.jmoldx.2020.05.004
- Mirzakhani M, Tarrahi MJ, Baghersad A, et al. Can couples With MCV≥80, MCH<26, HbA2 < 3.2, HbF<3 be classified as Low-risk β-thalassemia group? J Pediatr Hematol Oncol. 2019;41(4):303–306. doi:10.1097/MPH.0000000000001423
- Wu Y, Yao Q, Zhong M, et al. Genetic research and clinical analysis of deletional Chinese Gγ+(Aγδβ)0 -thalassemia and Southeast Asian HPFH in South China. Ann Hematol. 2020;99(12):2747–2753. doi:10.1007/s00277-020-04252-7
- Chan NCN, Wong THY, Cheng KCK, et al. An evaluation for the causes of reduced Hb A2and the molecular characterization of HBD variants in Hong Kong. Hemoglobin. 2021;45(6):387–391. doi:10.1080/03630269.2021.1965619
- Wenger AM, Peluso P, Rowell WJ, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019;37:1155–1162. doi:10.1038/s41587-019-0217-9
- Viprakasit V, Ekwattanakit S. Clinical classification, screening and diagnosis for thalassemia. Hematol Oncol Clin North Am. 2018;32:193–211. doi:10.1016/j.hoc.2017.11.006
- Chen M, Ye Y, Liao L, et al. Hereditary spherocytosis overlooked for 7 years in a pediatric patient with β-thalassemia trait and novel compound heterozygous mutations of SPTA1 gene. Adv Hematol. 2020;25(1):438–445. doi:10.1080/16078454.2020.1846874.
- Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. NEJM. 2011;364:710–718. doi:10.1056/NEJMoa1010174
- Sakorn P, Nur-Afsan S, Monthathip T, et al. Hematological analysis in Thai samples With deletional and nondeletional HbH diseases. Lab Med. 2018;49(2):154–159. doi:10.1093/labmed/lmx068.
- Chen P, Li SQ, Wu H. Alpha 2 codon 30 deletion (deltaGAG) causing non-deletional hemoglobin H disease in guangxi province. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21(5):435–439. doi:10.3760/j.issn:1003-9406.2004.05.005.
- Ma SK, Chan AY, Chiu EK, et al. Haemoglobin H disease due to (SEA) α-globin gene deletion and α2-codon 30 (ΔGAG) mutation: a family study. Clin Lab Haematol. 2001;23(5):325–327. doi:10.1046/j.1365-2257.2001.00411.x
- Chen X, Luo M, Pan L, et al. A novel 4.9 Kb deletion at beta-globin gene is identified by the third-generation sequencing: case report from baoan, China. Int. J. Biochem. Mol. Biol. 2022;529:10–16. doi:10.1016/j.cca.2022.01.024.