
ABSTRACT
The two most common systemic amyloidosis types are immunoglobulin light chain (AL) and amyloid transthyretin (ATTR) amyloidosis, in which the precursor proteins responsible for amyloidosis are light chain and transthyretin, respectively. Identification of precursor proteins is paramount to determine the type of amyloidosis, given that both amyloidosis types lack specificity in clinical presentation. Congo red staining followed by immunohistochemistry or immunofluorescence using fibril protein-specific antibodies is crucial for the diagnosis of amyloidosis. Here we describe a patient who was initially diagnosed with AL amyloidosis due to strong positive kappa light chain staining results. However, the diagnosis was corrected to hereditary ATTR amyloidosis using mass spectrometry and gene sequencing, confirming the important role of mass spectrometry in identifying the amyloid precursor protein and ruling out false-positive result from immunohistochemistry.
Introduction
Amyloidosis is a multisystem disease that affects the heart, nervous system, skin, kidneys, liver, lungs and intestines. The most common systemic amyloidosis types are immunoglobulin light chain (AL) and amyloid transthyretin (ATTR) amyloidosis [Citation1]. To date, 36 amyloid fibril proteins have been reported to be associated with human amyloidosis and immunoglobulin light chain and TTR are the precursor proteins for AL and ATTR amyloidosis, respectively [Citation2].
In this case report, we present a patient who was initially diagnosed with AL amyloidosis due to strong light chain immunostaining by immunohistochemistry (IHC) results but later the diagnosis was corrected to ATTR amyloidosis. This highlights the paramount role of clinical suspicion and evaluation with mass spectrometry(MS) to accurately identify the amyloidogenic precursor protein.
Case presentation
A 63-year-old man first experienced fatigue 3 years ago. Subsequently, he developed gradual symmetrical numbness and reduced sensation of pain and temperature in all extremities, which worsened over time. Ten months later, he successively visited orthopaedics, neurology and vasculocardiology departments in the first hospital owing to the aggravation of above mentioned symptoms. Cerebrospinal fluid analysis showed no abnormalities and electromyography revealed damage to both motor and sensory axons of peripheral nerves in the extremities. Magnetic resonance imaging revealed lumbar spinal stenosis. Laboratory tests revealed N-terminal pro B type natriuretic peptide (NT-ProBNP) and cardiac troponin T (cTnT) levels of 398 pg/mL and 0.048 ng/mL, respectively. Coronary angiography revealed coronary atherosclerosis. Echocardiography revealed an interventricular septum thickness of ∼13 mm and left ventricular ejection fraction of 68%. Electrocardiography showed sinus rhythm with ST-T changes. He was diagnosed with peripheral neuropathy of unknown cause, lumbar spinal stenosis and hypertrophic cardiomyopathy. However, his symptoms were not significantly alleviated despite the administration of lipid-lowering agents and mecobalamin tablets for 3 months. He underwent echocardiography in a second hospital, which revealed an interventricular septum thickness of 17 mm. Pathologic evaluation of myocardial biopsy specimen demonstrated myocardial amyloid precipitates in myocardial interstitium, which were positive by Congo red staining and exhibited apple-green birefringence under polarised light microscopy ( A,B), suggesting amyloidosis. IHC analysis of the myocardial tissue revealed focal interstitial kappa light chain deposition and negativity for lambda light chain in the absence of M protein in the blood and urine. Moreover, serum free light chain of the patient was normal. Furthermore, the patient refused receiving further treatment due to uncertain diagnosis.
Figure 1. Pathologic evaluation of myocardial biopsy specimen and abdominal fat pad biopsy specimen A. Congo red staining showing focal amyloid deposition in interstitium of myocardial biopsy specimen(×400). B. Amyloid deposition in myocardial biopsy specimen exhibits apple-green birefringence under polarised light microscopy(×400).C. Congo red staining showing focal amyloid deposition in interstitium of abdominal fat pad biopsy specimen(×400). D. Amyloid deposition exhibits apple-green birefringence under polarised light microscopy(×200). E, F. IHC staining showing positivity for kappa light chain and negativity for lambda light chain(×400).
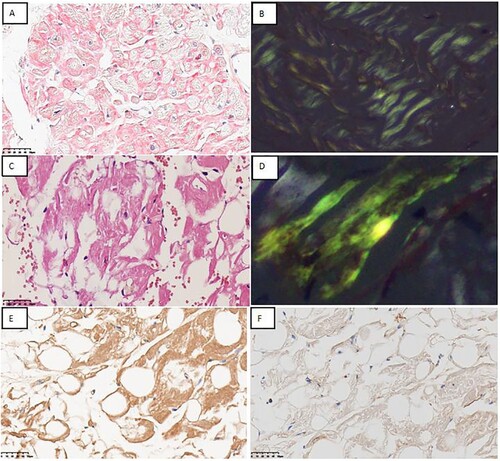
Ten months later, he was admitted to third hospital. His laboratory test results were as follows: NT-ProBNP, 682.2 pg/mL; cTnT, 0.054 ng/mL; serum β2-microglobulin, 2.73 Ug/mL; urine kappa free light chain (FLC), 29.9 mg/L and urine lambda FLC, 6.45 mg/L. Further, serum and urine immunofixation electrophoresis (IFE) and urinary protein electrophoresis results were negative. Urinary Bence–Jones protein, 24-h urinary total protein, 24-h urinary albumin and 24-h urinary FLC levels were normal. Notably, no evidence of clonal plasma cells was found in bone marrow morphology and biopsy. Congo red staining of the bone marrow biopsy specimen was negative. Abdominal fat pad biopsy specimen was positive by Congo red staining and exhibited apple-green birefringence under polarised light microscopy (C,D). The specimen was positively stained for kappa light chain in mesenchyme and negatively stained for lambda light chain ( E,F). Further, owing to the IHC positive results, he was diagnosed with stage 1 systemic AL amyloidosis. Subsequently, he received three cycles of chemotherapy with CyborD regimen (bortezomib 2.2 mg subcutaneous injection; cyclophosphamide 300 mg PO; dexamethasone 40 mg PO, at days 1, 8, 15, 22,and 35 days as one cycle). However, he experienced deteriorating of numbness in all four extremities since the start of chemotherapy.
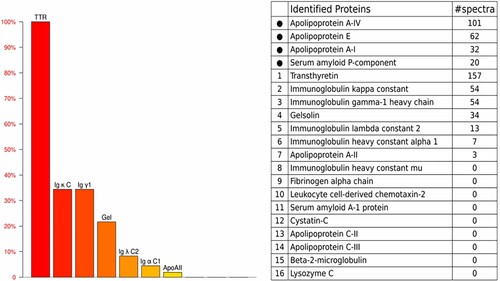
In June 2022, the patient presented to our department with serious numbness of extremities with suspicion of systemic amyloidosis. The results of laboratory tests performed at our centre were as follows: serum kappa FLC, 33.60 mg/L; serum lambda FLC, 16.70 mg/L; urinary kappa FLC, < 3.53 mg/L; urinary lambda FLC, < 2.1 mg/L; NT-ProBNP, 678 pg/mL; and cTnT, 0.055 ng/mL. Echocardiography scan revealed an interventricular septum thickness of 17 mm. Histopathologic examination of the bone marrow revealed that Congo red staining was negative. Further, IHC staining revealed that ∼0.26% of all nuclear cells were plasma cells, which had a kappa/lambda ratio of 1.24. Transthyretin amyloid cardiomyopathy (ATTR-CM) was suspected owing to the absence of M protein and negatively clonal plasma cell. Subsequently, as expected, 99mTc-pyrophosphate scintigraphy revealed grade 3 cardiac uptake. Notably, proteomic analysis by laser microdissection/mass spectrometry (LMD/MS) of abdominal fat amyloid revealed that the pathogenic protein was transthyretin (). However, genetic testing revealed that both patient and his son harboured the same mutation of Ala97Ser (p.Ala117Ser). Therefore, the final diagnosis was hereditary transthyretin (ATTRv) amyloidosis, and the patient was prescribed oral tafamidis 61 mg daily; he has not exhibited any adverse events to date.
Figure 2. LMD/MS analysis of abdominal fat pad biopsy specimen. The amyloid deposits demonstrated the highest relative abundance for TTR. LMD, laser microdissection ;MS, mass spectrometry; TTR, transthyretin; Igκ C, Immunoglobulin kappa constant ; Igγ1, immunoglobulin gamma-1 heavy chain; Gel, gelsolin; Ig λ C2, immunoglobulin lambda constant 2; Igα C1,immunoglobulin alpha constant 1; Apo, apolipoprotein.
Discussion
Congo red staining is the first step for diagnosing amyloid. However, a series of study have confirmed that CR staining of amyloids could make false positives, mainly due to its inherent limitations and characteristics [Citation3]. In particular, it is important to accurately identify the amyloidogenic precursor protein for subsequent treatment. Currently, the identification of amyloidogenic precursor proteins is mainly based on three methods: immunohistochemistry, mass spectrometric analysis and genetic testing. However, Each of these methods has both advantages and disadvantages. The immunohistochemical method is relatively applicable and simple in most situations, but the rate of false negative results is high [Citation4]. Conversely, our patient was misdiagnosed with AL amyloidosis based on false positive immunohistochemical results at the third hospital. To the best of our knowledge, a similar report does not exist in the literature. Unfortunately, we could not explain the underlying cause of false positive result. However, we confirmed that Congo red-positive and immunohistochemical staining results are insufficient to accurately diagnose amyloidosis in some special case.
Congo red staining followed by IHC or immunofluorescence using fibril protein-specific antibodies is critical for diagnosing amyloidosis and confirming fibril protein type. However, the assessment of amyloid by IHC is qualitative, non-standard and requires operator expertise [Citation5, Citation6]. Many antibodies associated with IHC lack specificity, occasionally resulting in simultaneous positivity for ATTR and immunoglobulin light chains (kappa or lambda light chains) [Citation7]. In addition, currently available antibodies are not applicable to rare forms of amyloidosis [Citation4]. Diagnosis of amyloidosis is a great challenge for clinicians owing to the complex multisystem manifestations of the disease. Similarly, our patient successively visited four different departments and was diagnosed with peripheral neuropathy, lumbar spinal stenosis and cardiomyopathy due to unknown cause. In such clinical scenarios, a multidisciplinary team approach is indubitably superior to a single disciplinary approach.
Another important issue that we cannot ignore is the use of correct diagnostic algorithm. Our patient underwent cardiac biopsy at the second hospital, and finally underwent scintigraphy and mass spectrometry at our hospital. According to the recommendations made by the Working Group on Myocardial and Pericardial Disease, when cardiac amyloidosis is suspected, 99mTc-PYP/DPD/HMDP scintigraphy and M protein assessment can be performed first. Further diagnostic strategy depends on the findings of these tests[Citation8].
99mTc-PYP/DPD/HMDP may be useful in grade 2–3 myocardial radiotracer uptake in ATTRv amyloidosis, whereas in AL amyloidosis, it is always in grade 0–1 myocardial radiotracer uptake [Citation9]. The results of cardiac uptake in our patient revealed grade 3 uptake, and this finding is consistent with ATTR cardiac amyloidosis.
Vrana et al. [Citation10]. reported that laser microdissection and mass spectrometry (LMD/MS) successfully identified amyloid types in 40 of 41 cases (98%) and that IHC provided information on fibril protein type in 15 of 36 cases (42%). When both methods were informative, the concordance between the results of LMD/MS and IHC was 100%. In the retrospective analysis of 320 samples, where IHC was used for amyloid identification, the amyloid type detected by LMD/MS agreed with IHC results in 91% of the samples. Additionally, the amyloid fibril protein was identified by LMD/MS in 80% (255 of 320) cases that could not be typed by IHC[Citation7]. Based on these results, LMD/MS has been proposed as the gold standard for the diagnosis of amyloidosis [Citation4].
TTR gene sequencing is mandatory in cases of highly suspicious ATTRv amyloidosis based on the clinical presentation and imaging findings [Citation11]. Further, ATTRv amyloidosis can be excluded if TTR genetic testing reveals no mutations [Citation12]. Notably, a multiracial Southeast Asian cohort study examining ATTRv reported Ala97Ser (p.Ala117Ser) as the most common variant identified in 14 patients, accounting for 66.7% of the Chinese patients and 48.3% of the entire cohort. Parental history was positive in all patients. Patients with Ala97Ser (p.Ala117Ser) mutation had an early disease course (age < 50 years) and presented with neurological dysfunction as the first symptom, and they developed cardiac symptoms 2–9 years later [Citation13, Citation14]. Another single-centre retrospective study reported that the Ala97Ser mutation was the most common cause of ATTRv in southern China [Citation15]. Similarly, the present case, originating from southern China, carried the Ala97Ser mutation. In amyloidosis, false-positive results by immunohistochemical staining are rare, and this condition cannot be only confirmed by IHC after Congo red staining. Therefore, it is important to further identify amyloidogenic precursor proteins that lead to amyloidosis through different methods, such as gene sequencing and LMD/MS. However, LMD/MS is expensive and time-consuming, which is not a routine in clinical practice. In addition, LMD/MS requires manual and subjective interpretation of mass spectrometric data by an expert [Citation16]. Therefore, we look forward to more accurate and simpler detection methods in clinical practice in the future.
Statement of Ethics
This study protocol was reviewed and approved by the committee name and affiliation of Shenzhen Luohu Hospital Group, Luohu People's Hospital.
Consent
The patient provided written informed consent for the publication of this case, including the publication of images.
Acknowledgements
The authors thank this patient for his cooperation in this study. Author Contributions: Jiao Chen was responsible for project progress, collection of data and writing of the manuscript. Haifei Chen was responsible for project design, overall planning. Lingyun Zhou, Danbo Liu, Fang Du and Hongxian Xiang were responsible for patient management.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability Statement
All data generated or analysed during this study are included in this article. Further enquiries should be directed to the corresponding author.
References
- Gertz MA, Dispenzieri A. Systemic amyloidosis recognition, prognosis, and therapy: a systematic review. JAMA. 2020 Jul 7;324(1):79–89. doi:10.1001/jama.2020.5493. PMID: 32633805.
- Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020 Dec;27(4):217–222. doi:10.1080/13506129.2020.1835263. Epub 2020 Oct 26. PMID: 33100054.
- Yakupova EI, Bobyleva LG, Vikhlyantsev IM, et al. Congo Red and amyloids: history and relationship. Biosci Rep. 2019 Jan 15;39(1):BSR20181415. doi:10.1042/BSR20181415. PMID: 30567726.
- Leung N, Nasr SH, Sethi S. How I treat amyloidosis: the importance of accurate diagnosis and amyloid typing. Blood. 2012 Oct 18;120(16):3206–3213. doi:10.1182/blood-2012-03-413682. Epub 2012 Sep 4. PMID: 22948045.
- Aoki M, Kang D, Katayama A, et al. Optimal conditions and the advantages of using laser microdissection and liquid chromatography tandem mass spectrometry for diagnosing renal amyloidosis. Clin Exp Nephrol. 2018 Aug;22(4):871–880. doi:10.1007/s10157-018-1533-y. Epub 2018 Jan 25. PMID: 29372474.
- Rezk T, Gilbertson JA, Mangione PP, et al. The complementary role of histology and proteomics for diagnosis and typing of systemic amyloidosis. J Pathol Clin Res. 2019 Jul;5(3):145–153. doi:10.1002/cjp2.126. Epub 2019 Apr 2. PMID: 30740936.
- Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020 Sep 23;21(1):198. doi:10.1186/s12875-020-01252-4. PMID: 32967612.
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur J Heart Fail. 2021 Apr;23(4):512–526. doi:10.1002/ejhf.2140. Epub 2021 Apr 7. PMID: 33826207.
- Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 2 of 2-diagnostic criteria and appropriate utilization. Circ Cardiovasc Imaging. 2021 Jul;14(7):e000030. doi:10.1161/HCI.0000000000000030. Epub 2021 Jul 1. PMID: 34196222.
- Vrana JA, Gamez JD, Madden BJ, et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009 Dec 3;114(24):4957–4959. doi:10.1182/blood-2009-07-230722. Epub 2009 Oct 1. PMID: 19797517.
- Bustamante JG, Zaidi SRH. Amyloidosis. [Updated 2022 Aug 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-.
- Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015 Sep;86(9):1036–1043. doi:10.1136/jnnp-2014-308724. Epub 2015 Jan 20. PMID: 25604431
- Chen Z, Koh JS, Saini M, et al. Hereditary transthyretin amyloidosis- clinical and genetic characteristics of a multiracial South-East Asian cohort in Singapore. J Neuromuscul Dis. 2021;8(4):723–733. doi:10.3233/JND-210656. PMID: 34024775.
- Wang S, Peng W, Pang M, et al. Clinical profile and prognosis of hereditary transthyretin amyloid cardiomyopathy: a single-center study in South China. Front Cardiovasc Med. 2022 Jun 27;9:900313. doi:10.3389/fcvm.2022.900313. PMID: 35833187.
- Du K, Li F, Wang H, et al. Hereditary transthyretin amyloidosis in mainland China: a unicentric retrospective study. Ann Clin Transl Neurol. 2021 Apr;8(4):831–841. doi:10.1002/acn3.51328. Epub 2021 Mar 19. PMID: 33739616.
- Palstrøm NB, Rojek AM, Møller HEH, et al. Classification of amyloidosis by model-assisted mass spectrometry-based proteomics. Int J Mol Sci. 2021 Dec 28;23(1):319. doi:10.3390/ijms23010319. PMID: 35008745.