
ABSTRACT
Background
Plasma cell leukemia (PCL) is a rare and aggressive plasma cell disorder, exhibiting a more unfavorable prognosis than multiple myeloma. PCL is classified into pPCL and sPCL. Recently, the IMWG has recommended new PCL definition criteria, which require the presence of ≥5% circulating plasma cells in peripheral blood smears. Due to its low incidence, research on pPCL and sPCL is limited.
Methods
We conducted a retrospective study and analyzed clinical and cytogenetic data of pPCL and sPCL patients. Overall survival (OS) and progression-free survival (PFS) were assessed by the Kaplan-Meier method, and survival distributions were compared using the log-rank test.
Results
This is a small cohort comprising 23 pPCL and 9 sPCL patients. Notably, sPCL patients showed a higher incidence of extramedullary infiltration and a higher percentage of bone marrow plasma cells (p = 0.015 and 0.025, respectively). Although no significant difference was found between the two groups in OS and PFS, a trend emerged suggesting a superior survival outcome for pPCL patients, with a higher cumulative 1-year PFS rate (38.3% vs. 13.3%) and a lower early mortality rate (mortality rate at 3 months: 15% vs. 33%). We also suggested that pPCL patients carrying t(11;14) may have a longer median survival time than individuals with other cytogenetic abnormalities, but this was not confirmed due to the small sample size.
Conclusion
Our study revealed clinical and cytogenetic features of pPCL and sPCL patients according to the new diagnostic criteria. The findings suggested a generally better prognosis for pPCL than sPCL and the likelihood of t(11;14) translocation acting as a favorable prognostic factor in pPCL. It is important to note that our study had a limited sample size, which may lead to bias. We hope well-designed studies can be conducted to provide more results.
Introduction
Plasma cell leukemia (PCL) is a rare and aggressive variant of plasma cell dyscrasia, characterized by plasmacytosis in peripheral blood (PB). PCL is classified as primary when it presents de novo in patients without previous evidence of multiple myeloma (MM) or secondary when it presents as a leukemic progression in patients with a history of MM. The crude incidence of primary plasma cell leukemia (pPCL) in Europe is 0.4 new cases per million [Citation1]. Among 75399 patients with a diagnosis of MM in the Surveillance, Epidemiology, and End Results (SEER) database from 1973 to 2009, PCL accounted for approximately 0.6% [Citation2]. From 1960 to 2008, of newly diagnosed MM patients seen at the Mayo Clinic, 1.3% were diagnosed with PCL [Citation3]. Generally, pPCL accounts for about 60–70% of all PCL cases, while secondary plasma cell leukemia (sPCL) constitutes the remaining 30–40% [Citation4,Citation5].
The original definition of PCL was based on the presence of an absolute count of plasma cells ≥2 × 109/l and/or ≥20% circulating plasma cells (CPCs) in PB. However, the International Myeloma Working Group (IMWG) consensus noted that the criteria were overly restrictive and resulted in missed diagnoses of PCL [Citation6]. Recently, some retrospective studies have already revealed a similar adverse prognostic impact of the presence of ≥5% CPCs as previously defined PCL [Citation7, Citation8]. Based on present evidence, the presence of 5% or greater CPCs on PB smears was considered a new criterion for pPCL diagnosis [Citation4]. However, this recommendation has not been widely used either in clinical or academic settings. Whether this revised definition applies to sPCL or not remains controversial.
Due to the relatively low incidence and prevalence of the disease, the genetic background and prognosis of the two types of PCL are not fully elucidated. Moreover, limited reported series have mainly focused on pPCL cases, and only a few sporadic sPCL patients have been reported. Herein, we compared biological characteristics, cytogenetic features, treatment response, and clinical outcomes between pPCL and sPCL under the new definition of ≥5% CPCs on PB smears.
Methods
Data source
This was a single-center retrospective study of patients diagnosed with PCL between January 2013 and December 2022 at Zhongda Hospital, China. PCL was defined by the presence of 5% or more CPCs on PB smears according to IMWG [Citation4]. Among PCL patients, 23 patients were diagnosed with pPCL and 9 patients developed sPCL. Laboratory and clinical data and treatment regimens were extracted from the electronic medical records. The following were collected: age at diagnosis of pPCL or sPCL, gender, first symptoms, paraprotein isotype, MM stage as defined by the Durie-Salmon staging system and international scoring system (ISS) [Citation9], karyotype, cytogenetic aberrations detected by fluorescence in situ hybridization (FISH) at the time of pPCL diagnosis or sPCL transformation. Additionally, hemoglobin level, platelet count, white blood cell count, levels of calcium, serum albumin, creatinine, β2-microglobulin and serum lactate dehydrogenase (LDH), incidences of extramedullary infiltration (EMI) and lytic lesions at diagnosis of pPCL or sPCL, treatment regimens, and therapeutic responses were collected. The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Zhongda Hospital, China (2023ZDSYLL043-P01).
Fluorescence in situ hybridization
Cytogenetic abnormalities were performed by FISH after the bone marrow plasma cells (BMPCs) were purified based on CD138 surface expression. The following probes were used in the panel: 13q14.3 (D13S319), 17p13.1 (TP53), 1q21(CKS1B), t(4;14)(p16;q32) FGFR3/immunoglobulin H (IgH), t(14;16)(q32;q23) IgH/MAF, t(11;14)(q13;q32) CCND1/IgH, and t(14;20)(q32;q12) IgH/MAFB. 1q gain describes patients with 3 copies of 1q, whereas 1q amplification denotes patients who have amplification of 1q, defined as 4 or more copies [Citation10]. The positive cut-off levels recommended by the European Myeloma Network (EMN) were defined as 10% for fusion or break-apart probes and 20% for deletions or numerical abnormalities [Citation11].
High-risk cytogenetic abnormalities (HRCA) were defined by detection of t(4;14), t(14;16), t(14;20), del(17p), p53 mutation or gain 1q, and standard-risk cytogenetic abnormalities were defined as including trisomies, t(11;14) or t(6;14) according to the modified risk stratification mSMART 3.0. Double hit myeloma was defined as having any 2 of the above HRCA [Citation12].
Treatment and efficacy assessment
The first-line treatment regimens patients received after diagnosis of pPCL or sPCL were divided into 4 types: proteasome inhibitor (PI)-based treatment (bortezomib or ixazomib), immunomodulatory drugs (IMiDs)-based treatment (thalidomide, lenalidomide or pomalidomide), PI + IMiDs-based treatment and conventional chemotherapy. No one in this cohort received carfilzomib as the first-line treatment. Treatment outcomes classified as complete response (CR), very good partial response (VGPR), partial response (PR), and no response (NR, both stable and progressive disease) were evaluated following the IMWG criteria [Citation13]. The objective response rate (ORR) was defined as CR, VGPR, or PR.
Statistical analysis
Descriptive statistics (median and range) were calculated for all continuous variables. Fischer’s exact test was performed to examine possible differences in categorical variables. Wilcoxon rank sum/Kruskal Wallis test or student’s t-test were used for continuous variables. For survival analyses, overall survival (OS) was calculated from diagnosis to death resulting from any cause or last follow-up. Progression-free survival (PFS) was measured from diagnosis until disease progression, relapse, or death. Survival curves were plotted using the Kaplan-Meier method, with comparison using the log-rank test. All statistical analyses were performed with SPSS 27 software (IBM, Armonk, NY) or by using the ‘survival’ and ‘survminer’ packages in R software 4.2.2 (R Foundation for Statistical Computing, Vienna, Austria). All analyses were two-sided and statistical significance was assumed at p < 0.05.
Results
Clinical characteristics of patients
The clinical data of 23 pPCL and 9 sPCL patients are summarized in . The median time of leukemic transformation for sPCL patients was 28 months (range: 13–92 months). There was no significant difference in baseline characteristics between pPCL patients and sPCL patients other than the percentages of BMPCs and EMI. A higher proportion of BMPCs was observed in sPCL with a median of 66.8% while the median in pPCL was 46.8% (p = 0.025), which is consistent with disease progression from MM to sPCL. The presence of EMI was more common in sPCL than in pPCL (77.8% vs. 26.1%, p = 0.015).
Table 1. Clinical and biological characteristics of patients with pPCL or sPCL.
The median age at diagnosis of both pPCL and sPCL patients was 63 years old. In our cohort, the proportion of females among pPCL patients was 52.2%, whereas males constituted 55.6% of the sPCL group. Immunoglobulin G (IgG) was the most detected M protein type in both groups, followed by the light chain subtype. ISS stage Ⅲ was mainly found in pPCL patients (63.6% in pPCL vs. 37.5% in sPCL) while the predominance of Durie-Salmon stage Ⅲ was found in both groups (95.7% in pPCL and 77.8% in sPCL). In terms of first symptoms, 43.5% of pPCL patients represented bone pain or fracture, which was also one of the common symptoms of MM. Extramedullary tumors were most prevalent in sPCL patients, with one having pulmonary involvement and one having testis involvement, while renal dysfunction or anemia was rare. Of note, pPCL patients tended to present with decreased platelet levels and elevated β2-microglobulin levels compared to sPCL patients, although no statistical difference was found.
Prevalence of cytogenetic abnormalities
Overall, a distinct cytogenetic profile was shown between pPCL and sPCL patients (). Del(13q) was the most frequently detected cytogenetic abnormality in pPCL (50%) while 1q21+ was the most common finding in sPCL (75%). The incidence of del(17p) was 10.5% in pPCL patients and 37.5% in sPCL patients, respectively. The prevalence of t(11;14) was mainly found in pPCL patients (46.7% in pPCL vs. 28.6% in sPCL, p = 0.648). There was a significant statistical difference between patients with different numbers of HRCA in the two groups (p = 0.042). Notably, over 60% of sPCL patients were diagnosed with double-hit myeloma while its counterpart had a diagnosis rate of only 16.7%. Contrarily, no obvious differences were observed between the two types of PCL in risk stratification and karyotype, which is probably due to the limited number of patients in both groups.
Table 2. Cytogenetic characteristics of patients with pPCL or sPCL.
Survival outcomes
With the median follow-up time of the entire cohort being 7 months (interquartile range: 3–12.75 months) from pPCL or sPCL diagnosis, 14 (60.9%) pPCL and 8 (88.9%) sPCL patients died. In terms of the overall survival outcome, there was no significant difference between the two groups both in OS and PFS (OS: p = 0.27; PFS: p = 0.14). The median OS for pPCL and sPCL were 9 and 10 months, and the median PFS for pPCL and sPCL were 9 and 6 months, respectively (A and B). The cumulative 1-year OS rates were 38.8% (95%CI, 0.213–0.710) and 26.7% (95%CI, 0.083–0.858) for pPCL and sPCL patients, and the cumulative 1-year PFS rates for pPCL and sPCL patients were 38.3% (95%CI, 0.204–0.719) and 13.3% (95%CI, 0.022–0.817), respectively.
Figure 1. Survival outcomes of pPCL and sPCL. Overall survival (OS) (A) and progression-free survival (PFS) (B) between pPCL and sPCL patients. (C) Mortality rates of pPCL and sPCL at 1, 3, 6, 12, and 24 months after diagnosis were shown.
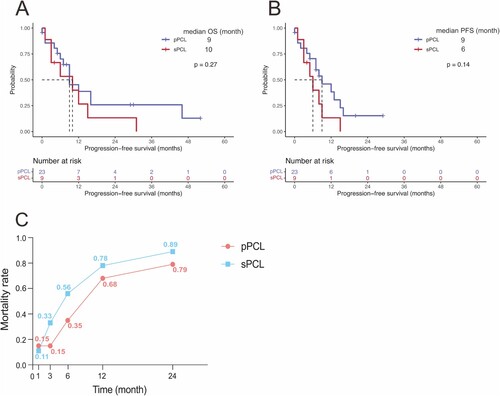
Similarly, patients with sPCL showed a higher early mortality rate than pPCL patients, especially within 3 months after diagnosis. Patients who died in the early stages of sPCL accounted for 11%, 33%, 56%, 78%, and 89% at 1, 3 months, 6 months, 12 months, and 24 months after diagnosis, respectively (C).
Treatment and efficacy
Treatment data were available for the entire cohort, except for 2 patients who refused medical treatment. Among 30 patients, response data of first-line therapies were available for 27 patients. The most used regimen in 21 pPCL patients who received treatment was VCd (bortezomib, cyclophosphamide, and dexamethasone), followed by PAd (bortezomib, doxorubicin, and dexamethasone) (A). Among these patients, 12 (57%) patients were treated with PI-based treatment, 4 (19%) patients received IMiDs-based therapy, 4 (19%) patients underwent PI + IMiDs treatment, and 1(5%) patient was treated with conventional chemotherapy. There were 4 patients undergoing autologous stem cell transplantation (ASCT) and none of them experience recurrence. In terms of sPCL, most patients received a minimum number of 2 MM treatment lines before sPCL diagnosis. Regimens for sPCL patients were heterogeneous, including VCd, Vd-PACE (bortezomib, dexamethasone, cisplatin, doxorubicin, cyclophosphamide, and etoposide), RCd (lenalidomide, cyclophosphamide, and dexamethasone), Pd (pomalidomide and dexamethasone) and so on (B). Only one person underwent ASCT after being diagnosed with sPCL, which could be related to the fact that the condition of sPCL was too poor to meet the requirements for ASCT. For first-line treatment of sPCL, there were 5 (56%) patients treated with PI-based regimens, 2 (22%) patients treated with IMiDs-based regimens, and 2 (22%) patients treated with conventional chemotherapy in sPCL. No sPCL patient underwent combination therapy of PI and IMiDs.
Figure 2. First-line treatment regimens and treatment response of pPCL and sPCL patients. The first-line treatment regimens for pPCL patients (A) and sPCL patients (B). (C) Overall objective response rate (ORR) in pPCL and sPCL patients, as well as ORRs in pPCL and sPCL patients treated with PI-based therapy or IMiDs-based therapy. Vd: bortezomib and dexamethasone, VCd: bortezomib, cyclophosphamide, and dexamethasone, PAd; bortezomib, doxorubicin, and dexamethasone, DVd: Daratumumab, bortezomib, and dexamethasone, Td: thalidomide and dexamethasone, TCd: thalidomide, cyclophosphamide, and dexamethasone, RCd: lenalidomide, cyclophosphamide, and dexamethasone, RVd: lenalidomide, bortezomib, and dexamethasone, VTd: bortezomib, thalidomide, and dexamethasone, VAd: vincristine, doxorubicin, and dexamethasone, Vd-PACE: bortezomib, dexamethasone, cisplatin, doxorubicin, cyclophosphamide, and etoposide, Pd: pomalidomide and dexamethasone, VMd, vincristine, mitoxantrone, and dexamethasone.
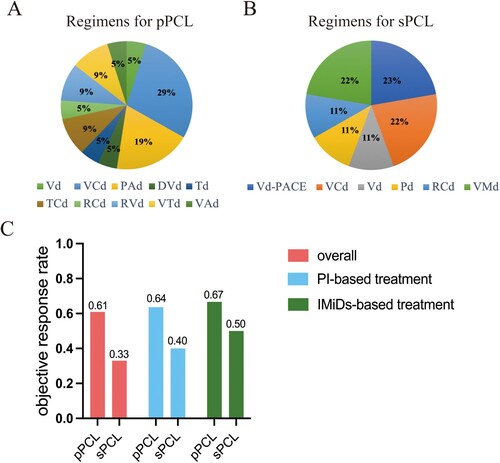
There was no significant difference in the overall ORR between pPCL and sPCL patients who received treatment (61% vs. 33%, p = 0.059). For patients undergoing PI-based treatment, ORR in pPCL and sPCL patients were 64% and 40%, respectively. In terms of IMiDs-based treatment, ORR in pPCL and sPCL patients were 67% and 50%, respectively (C). As is shown, the figures of ORR in pPCL patients are higher than that in sPCL, regardless of treatment regimens. Although there was no significant statistical difference between the two PCL groups due to the limited sample numbers, we supposed pPCL patients tended to have a potentially better response to anti-tumor therapy.
For patients treated with PI-based chemotherapy, the two kinds of PCL shared a similar prognosis. There was no significant difference in OS and PFS between the two groups (median OS: 9 months in pPCL vs.10 months in sPCL, p = 0.52; median PFS: 9 months in pPCL vs.8 months in sPCL, p = 0.39). In the IMiDs-based regimens cohort, both median OS and PFS were 8 months in sPCL patients, compared with 16 and 14 months in pPCL patients (OS: p = 0.14; PFS: p = 0.63).
Prognostic value of cytogenetic abnormalities
Cytogenetic abnormalities revealed different prognostic profiles when comparing pPCL with sPCL. The results are summarized in . The median OS and PFS for pPCL were 12 months, and the median OS and PFS for sPCL were 15 months in del(13q) patients. In pPCL, both the median OS for patients with del(17p) and those with 1q21+ was 9 months. The median PFS for patients harboring del(17p) and those with 1q21+ were 7 and 8 months, respectively. When it comes to sPCL, patients carrying del(17p) had a median OS of 12 months, while those with 1q21+ had a median OS of 6.5 months. The median PFS for these patients were 9 and 5.5 months, respectively. In addition, both the median OS and PFS in t(11;14) pPCL was 12 months whereas its counterpart exhibited a median OS of 6.5 months and a median PFS of 5 months. However, no significant difference was found in each subgroup mentioned above, possibly due to the limited cases available for analysis. There was a trend that pPCL patients carrying t(11;14) might exhibit a longer median survival time compared to individuals with other cytogenetic abnormalities (median OS: 12 months in t(11;14), 9 months in del(17p), and 9 months in 1q21+), which was consistent with the cognition that t(11;14) was a standard-risk cytogenetic marker with a more favorable outcome than other aberrations. Unfortunately, no statistical difference was observed between t(11;14) pPCL and non-t(11;14) pPCL in this small cohort (p = 0.51). The distinct presence of HRCA at diagnosis between pPCL and sPCL did not affect their prognosis (median OS: 12 vs. 8 months, p = 0.085, median PFS: 12 vs. 7 months, p = 0.13). Similarly, when we compared the lifespan of high-risk patients, there was no statistical difference between pPCL and sPCL (OS: p = 0.095, PFS: p = 0.18).
Table 3. Cytogenetic abnormalities and prognosis of pPCL and sPCL patients.
Discussion
It is very important to distinguish between different diseases that share a common origin. For example, myelodysplastic syndromes (MDS) are considered as ‘preleukemia’ condition, as approximately 20–30% of individuals with MDS ultimately progress to acute myeloid leukemia (AML) [Citation14]. Patients with AML have conventionally received more intense therapies than those with MDS. Similarly, PCL is an aggressive plasma cell dyscrasia that possesses distinct clinical and genetic characteristics, setting it apart from MM. Establishing a clear disease definition for PCL is essential, as it allows PCL patients to receive more specific therapies and improve their overall prognosis. The level of CPCs is generally considered to be associated with survival in PCL, but the appropriate level for PCL diagnosis is challenging and remains disputed. The traditional definition of PCL (≥20% CPCs and ≥2 × 109/l absolute plasma cells) may be too strict that inadvertently misclassify patients as MM when their disease is more aggressive and behaves more similarly to PCL, therefore limiting accurate reporting of outcomes to the PCL entity. Hence, reassessment of the plasma cell count defining the boundary of PCL and MM has been advocated in the latest IMWG consensus in view of patients diagnosed with a lower cutoff of ≥5% CPCs and ≥0.5 × 109/l absolute plasma cells in PB have the same adverse prognosis.
Two separate studies from Mayo Clinic compared the survival of patients with 5-19% CPCs to those with traditional definitions of pPCL characterized by 20% or greater CPCs. These studies demonstrated similar OS outcomes between the two groups, thus providing validation for the feasibility of the new diagnostic criteria [Citation7, Citation15]. In one of the studies, researchers also noted that outcomes of those with ≥5% CPCs were much poorer when compared with MM patients who did not have CPCs at diagnosis (p < 0.001) [Citation7]. Another Chinese study reported 91 pPCL patients under the revised criteria and 67 previously defined pPCL patients at a diagnosis between 2000 and 2019. Clinical characteristics were similar except for younger median age in patients with ≥20% CPCs. No statistical difference was found in survival outcomes in individuals harboring 5–19% CPCs compared to those with ≥20% CPCs (median PFS: 16 vs. 20 months, p = 0.26; median OS: 30 vs. 31 months, p = 0.89) [Citation16].
The rarity of PCL, particularly sPCL which has been experiencing an increasing incidence, has resulted in limited studies about this specific condition [Citation17]. This is a small retrospective study, aiming to display and compare clinical and genetic features, as well as disease outcomes between primary and secondary PCL under the new definition. In terms of clinical characteristics, no significant difference was found between pPCL and sPCL except that sPCL patients had higher percentages of BMPCs and EMI. Most pPCL patients were diagnosed with ISS stage Ⅲ. Interestingly, pPCL patients tended to present with lower platelet levels and elevated β2-microglobulin levels. The EMI may be related to impaired expression of adhesion molecules in PCL tumor cells, including CD56, leukocyte function-associated antigen (LFA) 1, and very late antigen (VLA) 5 [Citation18, Citation19]. In contrast with our results, Bezdekova et al. found an increased level of platelets at the time of pPCL diagnosis when compared with sPCL (median: 129.5 × 109/l vs. 49.1 × 109/l, p = 0.004) [Citation19]. We supposed that the increased platelet level and decreased β2-microglobulin level of sPCL patients in our cohort were associated with the prior MM treatment. When it comes to cytogenetic features, most pPCL patients presented with del(13q) while 1q21+ was the most frequently finding in sPCL patients. The prevalence of t(11;14) was mostly detected in pPCL while double-hit myeloma mainly occurred in sPCL.
The prognosis of both pPCL and sPCL is disappointing, even in the era of novel agents. Real-world studies from Greek, America, and Latin America implicated a median OS ranging from 18 to 21.6 months in pPCL [Citation20–22]. A multicenter retrospective study containing 101 sPCL patients, reported a median OS of 4.2 months. Of them, three-fourths of patients received novel agents, including PI, IMiDs, and monoclonal antibodies [Citation23]. According to our findings, the median OS was 9 months for pPCL and 10 months for sPCL, respectively. The relatively inferior median OS observed in pPCL within our cohort might be attributed to the low ASCT utilization rate. It is important to note that the enrollment of only 9 sPCL patients in this study introduces a potential for selection bias, which may have influenced the observation differences in survival time. Although no significant difference was found between the two groups in OS, there was a noticeable trend that pPCL patients might have superior survival, with a higher cumulative 1-year PFS rate and a lower early mortality rate. The results of previous studies also corroborated our assumption. Tiedemannp et al [Citation24]. and Papadhimitriou et al [Citation25]. agreed that prognostic stratification was based on cytogenetic background. On one hand, the better survival of patients diagnosed with pPCL was strongly associated with the presence of t(11;14) translocations. On the other hand, sPCL had a higher number of FISH abnormalities.
The prognosis for PCL depends on the heterogeneity of cytogenetic background. As a consequence, cytogenetic abnormalities have been included in the prognostic evaluation systems [Citation12, Citation26]. In agreement with previous reports [Citation15, Citation27], our study suggested the likelihood of t(11;14) acting as a favorable prognostic factor with improved survival in pPCL. Gang An et al. also noticed that all the t(11;14) sPCL cases in their study cohort had at least one other genetic aberration when initially diagnosed as MM, which could partly explain the shorter median OS and PFS in sPCL patients with t(11;14) compared to pPCL patients with the same translocation [Citation28]. It is important to note that the number of enrolled pPCL patients is quite small, which could be responsible for the insignificant statistical difference in our study. Nowadays, novel therapy is very promising in patients carrying t(11;14) translocation. Preclinical studies revealed that t(11;14) translocation was mainly associated with sensitivity to venetoclax due to the expression of B-cell leukemia/lymphoma-2 (BCL-2) [Citation29, Citation30]. Venetoclax single-agent showed clinical activity in patients with t(11;14) MM, with an ORR rate of 40% and a ≥VGPR rate of 27% [Citation31]. Touzeau et al. also found the high sensitivity of samples from MM patients with t(11;14) to venetoclax, including one patient diagnosed with pPCL [Citation32]. In addition, t(11;14) relapsed or refractory MM patients receiving venetoclax in combination with other antimyeloma therapies (bortezomib or daratumumab) showed promising results in achieving deep remission. The proportion of patients achieving ≥CR in these studies ranged from 54% to 58% [Citation33,Citation34]. Currently, there are very little data from case reports regarding the efficacy of venetoclax in pPCL harboring t(11;14). All the patients mentioned in the case reports were treated with venetoclax after relapse, leading to the achievement of minimal residual disease (MRD)-negative response [Citation35–38]. A recent study of a total of 91 pPCL patients demonstrate that TP53 mutation was associated with a significantly lower PFS and OS (p < 0.05) [Citation39]. The finding from different retrospective studies was that the presence of del(13q), 1p deletion, and t(4;14) were unfavorable prognostic biomarkers in PCL [Citation40–42]. In contrast, some studies did not consider del(13q) and 1q21 amplification as prognostic factors [Citation24, Citation40]. In terms of karyotype, the hyperdiploid one was more frequently observed in sPCL, which was probably due to the evolution from hyperdiploid MM [Citation24, Citation42]. Overall, heterogeneity was observed among the study results. The inconsistency of findings between studies may be explained by the relatively small sample size. Therefore, well-designed and long-term follow-up studies are expected to further investigate the prognostic role of cytogenetic abnormalities in PCL, especially in sPCL.
In recent decades, the emergence of novel agents, such as IMiDs and PI have been breakthroughs in the treatment of MM as well as PCL, improving long-term survival [Citation2, Citation43]. A phase II trial conducted in Italy reported an ORR of 74% and a median OS of 28 months in a pPCL cohort receiving Rd (lenalidomide and dexamethasone) treatment [Citation44]. The Greek myeloma study group retrospectively analyzed 42 PCL patients and 29 of them received a bortezomib-based regimen. A significantly higher ORR (69% vs. 31%, p = 0.04) and prolonged OS (median OS: 13 vs. 2 months, p < 0.007) were observed in patients treated with bortezomib compared with those who received conventional therapies. This study also revealed a deeper response to bortezomib in individuals with pPCL with a median OS of 18 months in comparison with 2 months in sPCL patients (p < 0.001) [Citation45]. A multicenter study of 101 sPCL patients reported that individuals who received a PI, either alone or in combination with conventional chemotherapy, IMiD or both, had higher ORR than those treated with other regimens (p = 0.01) [Citation23]. In addition, Nandakumar et al. showed a CR rate of 40% in a retrospective cohort of patients diagnosed with PCL under the new definition and treated with PI + IMiDs. The response rate was even better in individuals undergoing ASCT after the PI + IMiDs induction regimens, with 58% of the patients reaching CR [Citation15]. Inconsistent with prior reports [Citation45,Citation46], no significant difference in OS and PFS between pPCL and sPCL patients treated with the same regimen was observed in our study, probably due to the limited patients enrolled in this study and the heterogeneous therapeutic protocols.
There are many other promising therapies in PCL treatment. An early study proved that pPCL patients undergoing ASCT were associated with better survival data, where the survival was longer than 3 years in 64% of patients [Citation47]. Additionally, there was evidence from 2 prospective, randomized clinical trials supporting the use of PI + IMiDs induction regimens, followed by ASCT, for newly diagnosed PCL. Significant differences were also observed in median OS and PFS between patients undergoing ASCT and those considered not eligible for ASCT [Citation44, Citation48]. Novel agents, such as CD38 antibody (daratumumab and isatuximab) and chimeric antigen receptor (CAR) T-cell therapy have been used in several cases of PCL, achieving promising effects [Citation49, Citation50]. Other anti-MM agents including BCL-2 inhibitor venetoclax and exportin-1 inhibitor selinexor are undergoing assessment [Citation51].
There were several potential limitations in our study. First, this retrospective, single-center study enrolled relatively limited PCL cases and had some missing data. As a result, the small sample size may introduce bias in our results. Second, due to the limited patient number, we were unable to conduct univariate and multivariate analyses to further explore predictive biomarkers of the clinical outcome in PCL. Third, the therapeutic regimens in our study were relatively conventional. Few patients in our cohort received immunotherapy agents or targeted agents in such an era of novel agents.
In conclusion, our study is a valuable addition to the literature, as we presented the clinical and cytogenetic characteristics and their impacts on prognosis of pPCL and sPCL under the new definition. Although both pPCL and sPCL were aggressive disorders, a suggestion of superior survival in pPCL than sPCL was proposed. In addition, our findings suggested a trend of better survival outcomes in pPCL patients with t(11;14) translocation. Currently, large cohort clinical studies aiming at PCL, especially sPCL, are exceedingly scarce. With novel agents as the future of the therapeutic landscape, prospective studies in larger patient cohorts are expected to reveal the exact molecular background and further explore novel targeted drugs to improve the clinical course of PCL.
Authors contributions
J.G. collected and analyzed data and wrote the first draft of the manuscript. J.M reviewed the results and edited the manuscript. B.C. was responsible for supervising the data analysis. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Zhongda Hospital, China (2023ZDSYLL043-P01).
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Acknowledgments
The authors acknowledge the patients and physicians who participated in this project.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood. 2010;116:3724–3734. doi:10.1182/blood-2010-05-282632.
- Gonsalves WI, Rajkumar SV, Go RS, et al. Trends in survival of patients with primary plasma cell leukemia: a population-based analysis. Blood. 2014;124:907–912. doi:10.1182/blood-2014-03-565051.
- Gertz MA, Buadi FK. Plasma cell leukemia. Haematologica. 2010;95:705–707. doi:10.3324/haematol.2009.021618.
- de Larrea C F, Kyle R, Rosiñol L, et al. Primary plasma cell leukemia: consensus definition by the International Myeloma Working Group according to peripheral blood plasma cell percentage. Blood Cancer J. 2021;11:192. doi:10.1038/s41408-021-00587-0.
- Bladé J, Kyle RA. Nonsecretory myeloma, immunoglobulin d myeloma, and plasma cell leukemia. Hematol/Oncol Clin North Am. 1999;13:1259–1272. doi:10.1016/S0889-8588(05)70125-8.
- de Larrea C F, Kyle RA, Durie BGM, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013;27:780–791. doi:10.1038/leu.2012.336.
- Ravi P, Kumar SK, Roeker L, et al. Revised diagnostic criteria for plasma cell leukemia: results of a Mayo Clinic study with comparison of outcomes to multiple myeloma. Blood Cancer J. 2018;8:116. doi:10.1038/s41408-018-0140-1.
- Granell M, Calvo X, Garcia-Guiñón A, et al. Prognostic impact of circulating plasma cells in patients with multiple myeloma: implications for plasma cell leukemia definition. Haematologica. 2017;102:1099–1104. doi:10.3324/haematol.2016.158303.
- Greipp PR, San Miguel J, Durie BGM, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412–3420. doi:10.1200/JCO.2005.04.242.
- Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021;11:83), doi:10.1038/s41408-021-00474-8.
- Ross FM, Avet-Loiseau H, Ameye G, et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica. 2012;97:1272–1277. doi:10.3324/haematol.2011.056176.
- Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:1086–1107. doi:10.1002/ajh.26590.
- Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e346. doi:10.1016/S1470-2045(16)30206-6.
- Koeffler HP, Leong G. Preleukemia: one name, many meanings. Leukemia. 2017;31:534–542. doi:10.1038/leu.2016.364.
- Nandakumar B, Kumar SK, Dispenzieri A, et al. Clinical characteristics and outcomes of patients with primary plasma cell leukemia in the era of novel agent therapy. Mayo Clin Proc. 2021;96:677–687. doi:10.1016/j.mayocp.2020.06.060.
- Yan W, Fan H, Xu J, et al. The clinical characteristics and prognosis of patients with primary plasma cell leukemia (pPCL) according to the new IMWG definition criteria. Leuk Lymphoma. 2022;63:2955–2964. doi:10.1080/10428194.2022.2098290.
- Gundesen MT, Lund T, Moeller HEH, et al. Plasma cell leukemia: definition, presentation, and treatment. Curr Oncol Rep. 2019;21(1):8. doi:10.1007/s11912-019-0754-x.
- Moschetta M, Kawano Y, Sacco A, et al. Bone marrow stroma and vascular contributions to myeloma bone homing. Curr Osteoporos Rep. 2017;15:499–506. doi:10.1007/s11914-017-0399-3.
- Bezdekova R, Jelinek T, Kralova R, et al. Necessity of flow cytometry assessment of circulating plasma cells and its connection with clinical characteristics of primary and secondary plasma cell leukaemia. Br J Haematol. 2021;195:95–107. doi:10.1111/bjh.17713.
- Katodritou E, Terpos E, Delimpasi S, et al. Real-world data on prognosis and outcome of primary plasma cell leukemia in the era of novel agents: a multicenter national study by the Greek Myeloma Study Group. Blood Cancer J. 2018;8(3):31. doi:10.1038/s41408-018-0059-6.
- Usmani SZ, Nair B, Qu P, et al. Primary plasma cell leukemia: clinical and laboratory presentation, gene-expression profiling and clinical outcome with total therapy protocols. Leukemia. 2012;26:2398–2405. doi:10.1038/leu.2012.107.
- Peña C, Riva E, Schutz N, et al. Primary plasma cell leukemia in Latin America: demographic, clinical, and prognostic characteristics. A study of GELAMM group. Leukemia Lymphoma. 2023;64:816–821. doi:10.1080/10428194.2023.2171266.
- Jurczyszyn A, Castillo JJ, Avivi I, et al. Secondary plasma cell leukemia: a multicenter retrospective study of 101 patients. Leukemia Lymphoma. 2019;60:118–123. doi:10.1080/10428194.2018.1473574.
- Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–1052. doi:10.1038/leu.2008.4.
- Papadhimitriou SI, Terpos E, Liapis K, et al. The cytogenetic profile of primary and secondary plasma cell leukemia: etiopathogenetic perspectives, prognostic impact and clinical relevance to newly diagnosed multiple myeloma with differential circulating clonal plasma cells. Biomedicines. 2022;10(2):209. doi:10.3390/biomedicines10020209.
- Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. JCO. 2015;33:2863–2869. doi:10.1200/JCO.2015.61.2267.
- Cazaubiel T, Leleu X, Perrot A, et al. Primary plasma cell leukemias displaying t(11;14) have specific genomic, transcriptional, and clinical features. Blood. 2022;139:2666–2672. doi:10.1182/blood.2021014968.
- An G, Xu Y, Shi L, et al.. t(11;14) multiple myeloma: a subtype associated with distinct immunological features, immunophenotypic characteristics but divergent outcome. Leukemia Res. 2013;37:1251–1257. doi:10.1016/j.leukres.2013.06.020.
- Bodet L, Gomez-Bougie P, Touzeau C, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. 2011;118:3901–3910. doi:10.1182/blood-2010-11-317438.
- Punnoose EA, Leverson JD, Peale F, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist Venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15:1132–1144. doi:10.1158/1535-7163.MCT-15-0730.
- Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of Venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130:2401–2409. doi:10.1182/blood-2017-06-788786.
- Touzeau C, Dousset C, Le Gouill S, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28:210–212. doi:10.1038/leu.2013.216.
- Costa LJ, Davies FE, Monohan GP, et al. Phase 2 study of Venetoclax plus carfilzomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Blood Adv. 2021;5:3748–3759. doi:10.1182/bloodadvances.2020004146.
- Bahlis NJ, Baz R, Harrison SJ, et al. Phase I study of Venetoclax plus Daratumumab and dexamethasone, with or without Bortezomib, in patients with relapsed or refractory multiple myeloma with and without t(11;14). JCO. 2021;39:3602–3612. doi:10.1200/JCO.21.00443.
- Jelinek T, Mihalyova J, Kascak M, et al. Single-agent Venetoclax induces MRD-negative response in relapsed primary plasma cell leukemia with t(11;14). Am J Hematol. 2019;94:E35–E37. doi:10.1002/ajh.25331.
- Gonsalves WI, Buadi FK, Kumar SK. Combination therapy incorporating Bcl-2 inhibition with Venetoclax for the treatment of refractory primary plasma cell leukemia with t (11;14). Eur J Haematol. 2018;100:215–217. doi:10.1111/ejh.12986.
- Nalghranyan S, Singh AP, Schinke C. The combination of venetoclax, daratumumab and dexamethasone for the treatment of refractory primary plasma cell leukemia. Am J Hematol. 2020;95(2):E34–E35. doi:10.1002/ajh.25676.
- Roy T, An JB, Doucette K, et al. Venetoclax in upfront induction therapy for primary plasma cell leukemia with t(11;14) or BCL2 expression. Leukemia Lymphoma. 2022;63:759–761. doi:10.1080/10428194.2021.2010065.
- Cazaubiel T, Buisson L, Maheo S, et al. The genomic and transcriptomic landscape of plasma cell leukemia. Blood. 2020;136:48–49. doi:10.1182/blood-2020-139340.
- Chang H, Qi X, Yeung J, et al. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leukemia Res. 2009;33:259–262. doi:10.1016/j.leukres.2008.06.027.
- Pagano L, Valentini CG, De Stefano V, et al. Primary plasma cell leukemia: a retrospective multicenter study of 73 patients. Ann Oncol. 2011;22:1628–1635. doi:10.1093/annonc/mdq646.
- Avet-Loiseau H, Daviet A, Brigaudeau C, et al. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myélome and the Groupe Français de Cytogénétique Hématologique. Blood. 2001;97:822–825. doi:10.1182/blood.V97.3.822.
- Cowan AJ, Green DJ, Kwok M, et al. Diagnosis and management of multiple myeloma: a review. JAMA. 2022;327:464–477. doi:10.1001/jama.2022.0003.
- Musto P, Simeon V, Martorelli MC, et al. Lenalidomide and low-dose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia. 2014;28:222–225. doi:10.1038/leu.2013.241.
- Katodritou E, Terpos E, Kelaidi C, et al. Treatment with bortezomib-based regimens improves overall response and predicts for survival in patients with primary or secondary plasma cell leukemia: Analysis of the Greek myeloma study group. Am J Hematol. 2014;89:145–150. doi:10.1002/ajh.23600.
- Wang H, Zhou H, Zhang Z, et al. Bortezomib-based regimens improve outcome of patients with primary or secondary plasma cell leukemia: a retrospective cohort study. TJH [Internet]. 2020;37(2):91–97. doi:10.4274/tjh.galenos.2019.2019.0254.
- Mahindra A, Kalaycio ME, Vela-Ojeda J, et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia. 2012;26:1091–1097. doi:10.1038/leu.2011.312.
- Royer B, Minvielle S, Diouf M, et al. Bortezomib, doxorubicin, cyclophosphamide, dexamethasone induction followed by stem cell transplantation for primary plasma cell leukemia: a prospective Phase II Study of the Intergroupe Francophone du Myélome. J Clin Oncol. 2016;34:2125–2132. doi:10.1200/JCO.2015.63.1929.
- Li C, Cao W, Que Y, et al. A phase I study of anti-BCMA CAR T cell therapy in relapsed/refractory multiple myeloma and plasma cell leukemia. Clin Transl Med [Internet]. 2021;11(3):e346. doi:10.1002/ctm2.346.
- Yang C, Jiang N, Zhang L, et al. Relapsed/refractory multiple myeloma-transformed plasma-cell leukemia successfully treated with daratumumab followed by autologous stem cell transplantation. Ther Adv Hematol. 2021;12:1-5. doi:10.1177/2040620721989578.
- Chim CS, Kumar SK, Orlowski RZ, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018;32:252–262. doi:10.1038/leu.2017.329.