
ABSTRACT
Objectives
In patients with acute promyelocytic leukemia (APL), additional chromosomal abnormalities (ACAs) are prognostic indicators. However, the clinical features of ACAs were not systematically reported in Chinese patients. Therefore, we enrolled a large cohort of APLs to demonstrate the clinical characteristics and prognostic value of ACAs.
Methods
268 patients with newly diagnosed APL with t(15;17)(q24;q21) were retrospectively enrolled, and their clinical characteristics and the predictive value of ACAs were assessed between patients with the presence and absence of ACAs.
Results
APL patients with and without ACAs did not differ significantly in their clinical features or treatment response and clinical outcomes like overall survival (OS) and disease-free survival (DFS). It appeared to be substantially associated with worse OS in APL patients with trisomy 8, which was the most common ACA, although DFS was unaffected. Interestingly, the presence of ACAs or trisomy 8 affected OS and DFS in the subgroup of patients aged ≥60 years; by contrast, ACAs had no effect on OS or DFS in any treatment subgroup (ATRA + ATO/RIF or ATRA + ATO/RIF + CH or ATRA + CH), except for the ATRA + ATO/RIF + CH treatment subgroup, where their impact on DFS was less favorable.
Conclusions
Our results suggested that OS and DFS were unaffected by ACAs. Nonetheless, in the subgroup of patients older than 60, the existence of ACAs or trisomy 8 appeared to impact OS and DFS negatively. Individuals with t(15;17) alone had a higher DFS and were more susceptible to ATRA + ATO/RIF + CH than individuals with t(15;17) ACAs.
Introduction
Acute promyelocytic leukemia (APL), a unique genetic and clinical variety of acute myeloid leukemia (AML), accounts for up to 10%–15% of newly diagnosed adult cases of AML [Citation1]. The PML::RARα fusion gene produces the oncogenic PML-RARα fusion protein as a result of t(15;17)(q24;q21), which is thought to act as the primary pathogenic factor in APL via dysregulation [Citation2–4]. PML::RARα is the driving oncogene in APL and is responsible for the maturation arrest at the promyelocytic stage [Citation4]. The disease was renamed APL with PML::RARα by the WHO in 2023 to distinguish it from variant APL and occult genetic abnormalities. The therapeutic advancements over the last decades dramatically improved the prognosis and decreased the disease’s mortality [Citation1, Citation5]. Some patients still recur repeatedly due to factors such as resistance to retinoic acid and arsenic acid, and the poor curative effect of some patients has become a challenging issue.
However, other concomitant mechanisms associated with additional chromosome abnormalities (ACAs) may affect survival as a potential pathogenic factor of APL. Therefore, whether such ACAs alterations give independent prognostic information separate from or in addition to that provided by the underlying chromosomal defect, there are conflicting data in the literature on this point [Citation6–11]. To confirm the role of ACAs, we retrospectively analyzed the characteristics and prognostic impact of ACAs in a series of newly diagnosed APL patients with t(15;17)(q24;q21).
Patients and methods
Patients and data collection
This retrospective analysis collected the clinical and laboratory data of consecutive APL patients with the translocation t(15;17)(q24;q21) who initially presented to the First Affiliated Hospital, Zhejiang University, between January 2016 and December 2021. Based on the 2018 World Health Organization (WHO) categorization criteria, the bone marrow morphology, cytogenetics, immunophenotype, and molecular biology investigation provided a conclusive diagnosis for each patient. The treatment regiments for oral all-trans retinoic acid (ATRA) and injected arsenic trioxide (ATO) or oral realgarindigo naturalis formula (RIF) (as the ATRA + ATO/RIF group) and ATRA + ATO plus chemotherapy (CH) (as the ATRA + ATO/RIF + CH group) regimens had been described previously [Citation12–14]. When ATRA was intolerant, adot methods were implemented [Citation15].
The deadline for follow-up was 31 May 2023. This study was carried out by the institutional policy regarding protecting patients’ personal information and received clearance from the ethical committee of the First Affiliated Hospital, College of Medicine, Zhejiang University (2023 Ethical approval No. 0579).
Cytogenetic analysis
At diagnosis, 24-hour unstimulated and direct cultured bone marrow (BM) samples were processed according to industry standards. R-banding stained chromosomes and karyotypes were reported according to the International System for Human Cytogenetic Nomenclature 2013. If sufficient t(15;17) cell division figures were not observed, but fluorescence in situ hybridization and/or fusion genetic test revealed PML::RARα positivity, it was permitted to be included in the data analysis. Based on cytogenetic data, patients were divided into two groups: those with a single t(15;17) as the alone group and others as t(15;17) with ACAs. Patients with der(17)t(15;17), der(15)t(15;17) were placed in the group t(15;17) with alone.
Definitions of treatment response and endpoints
Classification of risk: low-intermediate-risk patients were defined by a presenting white blood cell (WBC) count ≤10 × 109/L, and high-risk patients had a WBC count >10 × 109/L [Citation16]. Early death was defined as death within 30 days. Hematologic complete remission (CR) and relapse were determined according to International Working Group criteria [Citation17]. Overall survival (OS) time was defined as the period from the patient's precise disease diagnosis until death (including death from any cause), missing, or the end of follow-up. Disease-free survival (DFS) time was measured from the date of achievement of remission until the date of relapse or death from any cause; it was only used in analyses of patients who achieved complete remission.
Statistical analysis
SPSS 25.0 for Windows was used to conduct all statistical analyses. Continuous data were presented as median (interquartile range, IQR) as appropriate and compared using the Mann–Whitney non-parametric test. The chi-square or Fiher’s exact test was used to compare count data. Group difference was examined by the Kaplan-Meier survival analysis method and log-rank test. All R packages ‘survminer’ (v.0.4.9) and ‘survival’ (v3.4.0) were generated through Hiplot Pro (https://hiplot.com.cn/), a comprehensive web service for biomedical data analysis and visualization. Univariate and multivariate analyses were performed using the Cox proportional hazards regression model for OS and DFS. The criterion for statistical significance was p < 0.05, and all p values reported were 2-sided.
Results
Characteristics of chromosomal abnormalities
268 newly diagnosed APL patients with t(15;17) were cytogenetically investigated in the study. The t(15;17) was presented as the sole abnormality in 208 (208/268, 78%) of these 268 patients, and the t(15;17) with ACAs was observed in 60 (60/268, 22%). Karyotypes of the ACAs were shown in Supplementary Table 1. It was further divided into four groups based on the number of ACAs in the evaluable cohort of APL patients with t(15;17): no ACA, one ACA, two ACAs, three or more ACAs, and the frequencies were 78% (208/268), 13% (36/268), 6% (15/268), 3% (9/268) respectively. Trisomy 8 was the most frequently observed ACA or combined with other anomalies in 23 cases (23/60, 38%). The chromosomes of markers were present in the group with ACAs in 16 cases (16/60, 27%). In addition to the 4, 9, 10, and Y chromosomes, other secondary chromosomal alterations comprised structural and/or numerical aberrations.
Main clinical characteristics and response to therapy
In total, 7 patients were lost due to contact failure among the 268 patients with a median follow-up time of 47 months (range, 1–89 months), of which 3 instances were lost after hematological relapse. There was no significant difference in the main clinical characteristics and response to therapy between the groups with t(15;17) alone and t(15;17) with ACAs (). Similarly, no significant impact was observed in the clinical features and response to therapy, such as age, sex, white blood cells, and the rate of hematologic CR between patients with trisomy 8 and those with other ACAs (p > 0.05, Supplementary Table 2).
Table 1. Clinical characteristics and response to treatment of with t(15;17) APL patients.
Influence of ACAs on prognosis in APL
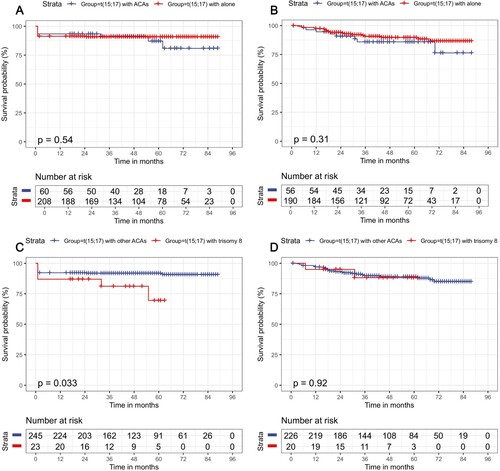
There was still no significant difference in OS (p = 0.54) and DFS (p = 0.31) (A and B) between APL patients with t(15;17) alone and with other ACAs. Interestingly, APL patients with trisomy 8 had a poorer OS compared with those with other ACAs (p = 0.033, C) but not DFS (p = 0.92, D). The OS and DFS were not affected among patients with 0–2 ACAs and those with 3 or more ACAs (p = 0.294, p = 0.283, respectively). Otherwise, no statistical difference was demonstrated for the incidence of OS (p = 0.587) and DFS (p = 0.203) among groups based on the number of ACAs besides t(15;17).
Figure 1. Survival outcomes for APL patients according to the presence of ACAs or trisomy 8. (A) OS in the ACAs group; (B) DFS in the ACAs group; (C) OS in the trisomy 8 group; (D) DFS in the trisomy 8 group.
Influence of ACAs combined with age or treatment on the prognosis of APL
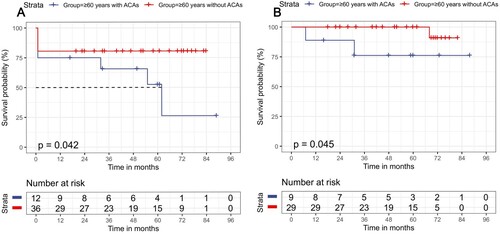
The patients with age ≥60 years were shown to have lower OS than the younger patients (p < 0.001, Supplementary Figure 1A) and unaffected DFS (p = 0.430, Supplementary Figure 1B). Compared with t(15:17) alone, the group of t(15;17) plus ACAs showed a slightly lower OS and DFS in the subgroup of patients aged ≥60 years (p = 0.042, p = 0.0.045, respectively, A and B). However, the OS and DFS of the two groups were not statistically significant in the subgroup of patients aged <60 years (p = 0.205, p = 0.735, respectively). Interestingly, t(15;17) with trisomy 8 showed worse OS and DFS than t(15:17) with other ACAs in the subset of patients with age ≥60 years (p = 0.012, p < 0.001, respectively, Supplementary Figure 1C and D).
Figure 2. Survival outcomes for APL patients in the subgroup of age ≥60 years. (A) OS in the patients with aged ≥60 years subgroup; (B) DFS in those with aged ≥60 years subgroup.
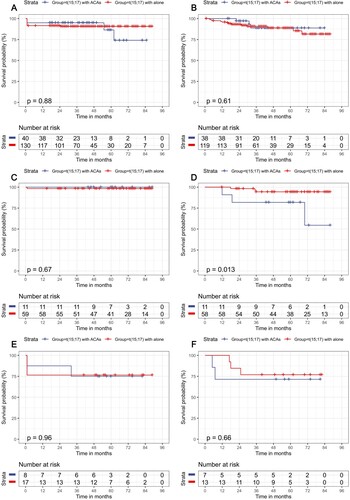
DFS was unaltered (p = 0.11), but OS differed between different treatment modalities in the remaining 265 patients (p = 0.022), except for three patients who died too soon after ATRA treatment initiation (Supplementary Figure 2A and B). The ATRA + ATO/RIF + CH therapy exhibited a better OS than either the ATRA + ATO/RIF subgroup (p = 0.015) or the ATRA + CH subgroup (p < 0.001). There was no difference in OS or DFS between patients with and without ACAs in the three therapy subgroups of ATRA + ATO/RIF, ATRA + ATO/RIF + CH, or ATRA + CH (all p > 0.05, Figure 3A–C, E and F), except that the DFS of the ATRA + ATO/RIF + CH cohort with ACAs was poorer than that of t(15; 17) alone (p = 0.013, D).
Figure 3. Survival outcomes for APL patients in various treatment subgroups according to the presence or absence of ACAs. (A) OS in the ATRA + ATO/RIF subgroup; (B) DFS in the ATRA + ATO/RIF subgroup group; (C) OS in the ATRA + ATO/RIF + CH subgroup; (D) DFS in the ATRA + ATO/RIF + CH subgroup; (E) OS in ATRA + CH subgroup; (F) DFS in ATRA + CH subgroup.
Additionally, Supplementary Table 3 analyzed the relationship between OS or DFS and several selected factors, including age, sex, karyotype (with or without ACAs), initial WBC level, and treatment. Age ≥60 years, initial WBC count and ATRA + ATO/RIF + CH therapy were identified as independent predictive factors of OS outcome among those selected (HR 4.02, 95% CI: 1.75–9.26, p = 0.001; HR 1.03, 95% CI: 1.02–1.03, p < 0.001; HR 0.06, 95% CI: 0.08–0.48, p = 0.009, respectively). The univariate study indicated a slight correlation between DFS and treatment with ATRA + ATO/RIF + CH (HR 0.30, 95% CI: 0.09–0.98, p = 0.045). However, multivariate analysis showed no significant impact from these components (HR 0.32, 95% CI: 0.10–1.05, p = 0.060). Neither OS nor DFS was affected by the factor ACAs. Likewise, trisomy 8 was shown by the Cox analysis to be a non-independent predictor of OS and DFS.
Discussion
The karyotypic results were consistent with earlier data for APL patients, in which 22% (60/268) of the patients had ACAs besides t(15;17) in this study, a slightly lower figure than that (22/73, 30%) found in previous studies [Citation18]. Consistent with previous results [Citation6, Citation7, Citation10, Citation11, Citation19], we found that ACAs beyond t(15;17) did not affect APL’s CR rate, early death, DFS, or OS. Furthermore, whether the t(15;17) with or without ACAs, 67% (179/268) patients in this study had at least one residual normal metaphase on karyotypes. The presence of residual normal metaphases had no prognostic significance on CR rate (p = 0.930) and OS (p = 0.136). Along with the previous report [Citation9], the study further supported the conclusion that residual normal metaphases did not confer predictive value, such as OS, in APL patients with t(15;17).
Whereas trisomy 8 was primarily represented in 38% (23/60) of patients with total ACAs, a little higher incidence than the previous studies [Citation18]. Trisomy 8 besides t(15;17) was observed to have a worse OS for APL. This contradicted previous reports [Citation6, Citation20]. It may be due to the limited data tested in retrospective analysis or racial reasons. Notably, in the subgroup of APL patients aged 60 years or older, the group t(15;17) with ACAs or trisomy 8 had a lower OS and DFS than alone. However, multivariate analysis revealed that age ≥60 was an adverse and independent prognostic factor for OS in APL. This suggested that older APL patients require special treatment, and the management of APL in elderly patients was also highlighted [Citation9].
In line with previous reports [Citation14, Citation21], this study showed that ATRA + ATO/RIF + CH was more effective on OS than ATRA + ATO/RIF in treating APL patients, contrary to previous findings [Citation22]. However, neither treatment affected DFS, and even in high-risk patients, no treatment affected OS (p = 0.057) or DFS (p = 0.849), which is consistent with the report [Citation22]. In the present study, the presence or absence of ACAs beyond t(15;17) did not affect OS in either ATRA + ATO/RIF or ATRA + ATO/RIF + CH or ATRA + CH treatment groups. In contrast to patients with t(15;17) ACAs, APL patients with t(15;17) alone had a higher DFS and were more susceptible to ATRA + ATO/RIF + CH. In short, many studies on this assertion are limited by relatively small sample sizes or varying treatment approaches at various stages of therapeutic development, leading to conflicting data regarding the prognostic implications of ACAs.
Future studies should gather more data by larger sample sizes with longer follow-up durations or multicenter conjoint analysis to corroborate these early findings and increase the statistical power to address the effect of ACAs or trisomy 8 or treatment on survival, for sure. If confirmed, treatment patterns may change.
Supplemental Material
Download MS Word (640 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/16078454.2024.2314398)
References
- Huynh TT, Sultan M, Vidovic D, et al. Retinoic acid and arsenic trioxide induce lasting differentiation and demethylation of target genes in APL cells. Sci Rep. 2019;9(1):9414. doi:10.1038/s41598-019-45982-7
- Rahimi N, Zhang Y, Mina A, et al. An integrative approach reveals genetic complexity and epigenetic perturbation in acute promyelocytic leukemia: a single institution experience. Hum Pathol 2019;91:1–10. doi:10.1016/j.humpath.2019.05.008
- Noguera NI, Catalano G, Banella C, et al. Acute promyelocytic leukemia: update on the mechanisms of leukemogenesis, resistance and on innovative treatment strategies. Cancers (Basel). 2019;11(10). doi:10.3390/cancers11111740
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544.
- de Thé H, Pandolfi PP, Chen Z. Acute promyelocytic leukemia: A paradigm for oncoprotein-targeted cure. Cancer Cell. 2017;32(5):552–560. doi:10.1016/j.ccell.2017.10.002
- De Botton S, Chevret S, Sanz M, et al. Additional chromosomal abnormalities in patients with acute promyelocytic leukaemia (APL) do not confer poor prognosis: results of APL 93 trial. Br J Haematol. 2000;111(3):801–806. doi:10.1111/j.1365-2141.2000.02442.x
- Ghaddar HM, Pierce S, Reed P, et al. Prognostic value of residual normal metaphases in acute myelogenous leukemia patients presenting with abnormal karyotype. Leukemia. 1995;9(5):779–782.
- Wiernik PH, Sun Z, Gundacker H, et al. Prognostic implications of additional chromosome abnormalities among patients with de novo acute promyelocytic leukemia with t(15;17). Med. Oncol. (Northwood, London, England). 2012;29(3):2095–2101. doi:10.1007/s12032-012-0251-7.
- Sanz MA, Fenaux P, Tallman MS, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133(15):1630–1643. doi:10.1182/blood-2019-01-894980
- Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The medical research council adult and children’s leukaemia working parties. Blood. 1998;92(7):2322–2333.
- Hernández JM, Martín G, Gutiérrez NC, et al. Additional cytogenetic changes do not influence the outcome of patients with newly diagnosed acute promyelocytic leukemia treated with an ATRA plus anthracyclin based protocol. A report of the Spanish group PETHEMA. Haematologica. 2001;86(8):807–813.
- Zhang X, Zhang H, Chen L, et al. Arsenic trioxide and all-trans retinoic acid (ATRA) treatment for acute promyelocytic leukemia in all risk groups: study protocol for a randomized controlled trial. Trials. 2018;19(1):476. doi:10.1186/s13063-018-2812-3.
- Zhu HH, Wu DP, Du X, et al. Oral arsenic plus retinoic acid versus intravenous arsenic plus retinoic acid for non-high-risk acute promyelocytic leukaemia: a non-inferiority, randomised phase 3 trial. Lancet Oncol. 2018;19(7):871–879. doi:10.1016/S1470-2045(18)30295-X
- Iland HJ, Collins M, Bradstock K, et al. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian leukaemia and lymphoma group (ALLG) APML4 study: a non-randomised phase 2 trial. Lancet Haematol. 2015;2(9):e357–e366. doi:10.1016/S2352-3026(15)00115-5
- O'Donnell MR, Tallman MS, Abboud CN, et al. Acute myeloid leukemia, version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Comprehensive Cancer Network. 2017;15(7):926–957. doi:10.6004/jnccn.2017.0116
- Li J, Chen L, Zhu H, et al. Retinoic acid and arsenic trioxide with or without chemotherapy for acute promyelocytie leukemia with different risk stratifications: a interim analysis of China APL 2012 Study - ScienceDirect.
- Creutzig U, Kaspers GJ. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2004;22(16):3432–3433. doi:10.1200/JCO.2004.99.116
- Ono T, Takeshita A, Iwanaga M, et al. Impact of additional chromosomal abnormalities in patients with acute promyelocytic leukemia: 10-year results of the Japan Adult Leukemia Study Group APL97 study. Haematologica. 2011;96(1):174–176. doi:10.3324/haematol.2010.030205
- Lou Y, Suo S, Tong H, et al. Characteristics and prognosis analysis of additional chromosome abnormalities in newly diagnosed acute promyelocytic leukemia treated with arsenic trioxide as the front-line therapy. Leuk Res. 2013;37(11):1451–1456. doi:10.1016/j.leukres.2013.07.030
- Wolman SR, Gundacker H, Appelbaum FR, et al. Impact of trisomy 8 (+8) on clinical presentation, treatment response, and survival in acute myeloid leukemia: a Southwest Oncology Group study. Blood. 2002;100(1):29–35. doi:10.1182/blood.V100.1.29
- Wang H, Cao F, Li J, et al. Arsenic trioxide and mannitol for the treatment of acute promyelocytic leukemia relapse in the central nervous system. Blood. 2014;124(12):1998–2000. doi:10.1182/blood-2014-04-568121
- Wang HY, Gong S, Li GH, et al. An effective and chemotherapy-free strategy of all-trans retinoic acid and arsenic trioxide for acute promyelocytic leukemia in all risk groups (APL15 trial). Blood Cancer J. 2022;12(11):158. doi:10.1038/s41408-022-00753-y