
ABSTRACT
Background:
Bernard–Soulier syndrome (BSS) is a rare inherited macrothrombocytopenia, usually autosomal recessive, which is characterized by prolonged bleeding, thrombocytopenia, and abnormally large platelets.
Methods:
For more than 6 years, we misdiagnosed a patient with BSS without an obvious bleeding tendency as having idiopathic thrombocytopenia purpura (ITP), prior to obtaining a genetic analysis. On admission, routine hematology showed a platelet count of 30 × 109/L and mean platelet volume (MPV) of 14.0 fL.
Results:
Whole-exome sequencing revealed two likely pathogenic heterozygous mutations (c.95_101del and c.1012del) in GP1BA. Flow cytometry analysis of platelet membrane glycoproteins indicated that the expression of GP1b was 0.28% of the normal level. Platelet aggregation tests indicated that platelet aggregation was inhibited by ristocetin- (1.7%), ADP- (14.5%), and arachidonic acid- (5.6%) induced platelet aggregation. A literature review identified reports on 53 mutations in the GP1BA gene in 253 patients, 29 mutations in the GP1BB gene in 90 patients, and 32 mutations in the GP9 gene in 114 patients.
Conclusion:
This case report describes two novel gene mutation sites that have not been reported previously, enriching understanding of the GP1BA mutation spectrum.
Introduction
Bernard–Soulier syndrome (BSS) is a rare inherited macrothrombocytopenia, usually autosomal recessive, with an incidence rate less than one per million [Citation1]. The clinical characteristics of BSS generally include prolonged bleeding, thrombocytopenia and abnormally large platelets [Citation2].
The molecular mechanism of BSS is a defect in the genes encoding GPIb-IX-V complex, a platelet receptor for von Willebrand factor (VWF). Impaired platelet structure and function subsequently lead to a bleeding tendency, abnormally large platelets, and decreased platelet count [Citation3]. The gene mutations that have been reported to cause BSS include GP1BA, GP1BB, and GP9 mutations. A previous study found that GP1BA, GP1BB, and GP9 gene mutations accounted for 28%, 28%, and 44%, respectively, among the 211 families with BSS studied [Citation4].
In this report, we describe a case of BSS diagnosed in a child at the Children's Hospital of Soochow University, Suzhou, China. Through genetic analysis, we discovered two novel mutations in the GP1BA gene. Moreover, we performed a literature review to introduce the clinical characteristics of BSS and emphasize the crucial role of gene detection in early identification and diagnosis, thereby enabling better treatment.
Case presentation
The propositus, a girl aged 6 years and 11 months, was the first of two children born to non-consanguineous parents. She had a younger brother. She was born by cesarean section at full term, breastfed after birth, and grew well. She was found to have thrombocytopenia at birth. She had bleeding gums without an obvious cause at age 10 months, and an intracranial hemorrhage due to a fracture of the occipital bone caused by trauma at age 2 years. Morphological examinations of a bone marrow biopsy at age 4 years and 4 months showed normal morphology, whereas an examination at age 6 years and 2 months, showed findings suggestive of idiopathic thrombocytopenic purpura (ITP) (more than 200 megakaryocytes in the whole smear, mainly granular megakaryocytes, with low numbers of platelets and cluster visibility). A review of her routine hematology showed that her platelet count was maintained at 30–70 × 109/L. She was diagnosed with ITP but did not receive any specific treatment. At the age of 6 years and 11 months, she was admitted to our hospital because of fever for 3 days, vomiting for 1 day, and thrombocytopenia. She had an episode of epistaxis provoked by vomiting. Physical examination revealed that she was fully conscious and had pharyngeal congestion. Chest auscultation revealed coarse breath sounds in both lungs, with no obvious dry or wet rales. Inspection of the skin of the whole body revealed no bleeding spots or rash. Her routine hematology results were: white blood cell count, 3.36 × 109/L; platelet count, 30 × 109/L; absolute neutrophil count, 2.13 × 109/L; absolute lymphocyte count, 0.86 × 109/L; and mean platelet volume (MPV), 14.0 fL. Nucleic acid testing was positive for influenza, which explained the fever and vomiting, and she was treated with peramivir 300 mg once daily. Carbazochrome sodium sulfonate 20 mg once daily was administered as a hemostatic agent, starting on admission. We noted that her platelet count had been low since infancy and that her MPV was large (16.0 fL; normal, 7.4–11.0 fL), which suggested an inherited macrothrombocytopenia. Therefore, we conducted genetic analyses of the patient and her parents and did not use glucocorticoids or human immunoglobulin (the first-line treatment of ITP). Three days after admission, her fever and vomiting were controlled and her platelet count had increased to 42 × 109/L. The patient was discharged and followed up as an outpatient.
Detection method of platelet aggregation function and flow cytometry
Platelet aggregation function: Platelet aggregation function was measured using light transmission aggregometry (LTA). Platelet-rich plasma (PRP) and platelet-poor plasma (PPP) were added to the colorimetric cup, and an inducer was added to PRP to induce platelet aggregation. The aggregation rate and transmittance of PRP and PPP were set to 0% and 100% respectively. With platelet aggregation, plasma turbidity decreased and transmittance increased, and the platelet aggregation rate was automatically recorded by the instrument. Measurement was performed using a Sysmex CS-5100 automated coagulation analyzer (Sysmex Corporation, Kobe, Japan).
Flow cytometry: First, a peripheral blood sample was centrifuged (200 × g for 5 min) to separate the supernatant. Next, 5 μL of CD41-PE, CD61-PE, and CD42B-PE were added to a 2-mL tube containing 3 µL of supernatant, then co-mixed with 50 μL of physiological saline. After incubating for 15 min in the dark, 600 μL of physiological saline was added to the tube, mixed well, and the product was analyzed using a Navios 10 colors, 3 laser flow cytometer (Beckman Coulter Inc., Brea, CA, USA).
Molecular genetic analyses and platelet function test
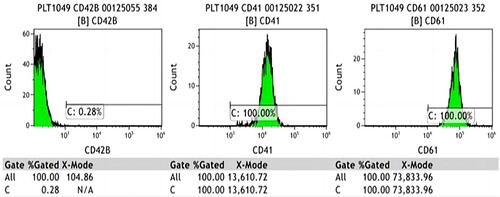
Whole-exome gene sequencing revealed three heterozygous mutations in the GP1BA gene of the proband: c.95_101del (p.Asn32Thrfs Ter5), c.1012del (p.Met338Cys fsTer62), and c.1616T>G (p.Phe539Cys). Sanger sequencing confirmed the existence of these mutations and revealed that the first variant was inherited from the father, and that the other two variants were inherited from the mother (). These three variants were not included in any databases, confirming them to be germline mutations. According to the criteria proposed by the American College of Medical Genetics and Genomics (ACMG) [Citation5], the c.95_101del mutation was classified as ‘likely pathogenic’ as evidenced by PVS1 and PM2_supporting (PVS1: the variant is a frameshift mutation that may result in loss of gene function; PM2_supporting: the variant was absent from the controls databases); the c.1012del mutation was also classified as ‘likely pathogenic’ as evidenced by PVS1 and PM2_supporting (PVS1: the variant is a frameshift mutation that may result in loss of gene function; PM2_supporting: the variant has the highest frequency of 0.0001 in the controls Database, and is not a polymorphic locus); whereas the c.1616T>G mutation was classified as ‘uncertain’ as evidenced by PM3 and PM2_supporting and BP4_Moderate. Alpha Fold was used to predict the protein structure of GP1BA (). Moreover, a missense mutation c.2303G>A(p.Arg768Gln) of VWF gene was detected in exon 18, which is ‘likely pathogenic’ according to the ACMG guidelines. However, in this case, it was demonstrated to be nonpathogenic because the antigen and VWF antigen activity were normal (154.0% and 215.1%, respectively). In combination with a compound heterozygous mutation in the GP1BA gene, the proband's phenotype is consistent with a diagnosis of BSS. After the results of genetic analyses were revealed, we conducted the platelet aggregation tests and flow cytometric analysis of platelet membrane glycoproteins of the patient and her parents. The platelet aggregation tests of the patient showed inhibition of ristocetin- (1.7%), ADP- (14.5%), and arachidonic acid- (5.6%) induced platelet aggregation. Flow cytometry analysis of platelet membrane glycoproteins indicated that the expression of GP1b was 0.28% of the normal level (). Platelet aggregation tests and flow cytometry analysis and flow cytometric analysis of platelet membrane glycoproteins of the patient's parents were normal, which indicated that these two mutations were autosomal recessive inheritance.
Figure 1. Sanger sequencing of GPI bα exons in the present case and her parents. (a) The patient had a heterozygous mutation of c.95_101del in chr17:4835994-4836000, father had a heterozygous mutation of c.95_101del, and mother had no mutation. (b) The patient had a heterozygous mutation of c.1012del in chr17:4836911-4836911, father had no mutation, and mother had a heterozygous mutation of c.1012del. (c) The patient had a heterozygous mutation of c.1616T>G in chr17:4837515, father had no mutation, and mother had a heterozygous mutation of c.1616T>G.
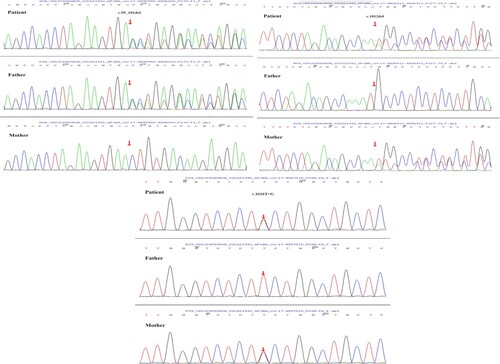
Figure 2. Protein structures of wild-type and mutant GP1BA. (PyMOL 2.5 software was used to generate 3D structural maps of GP1BA wild-type and mutant proteins.): (a) GP1BA wild-type protein, (b) c.95_101del mutant protein, (c) c.1012del mutant protein, (d) part of GP1BA wild-type protein, (e) part of c.1616T>G mutant protein.
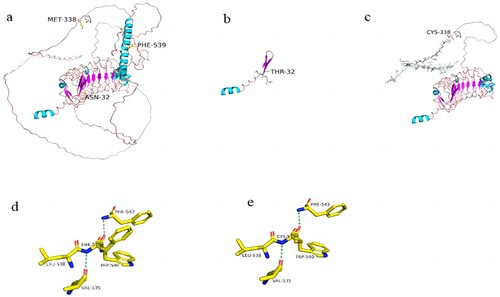
Figure 3. Flow cytometry analysis of GPIbα. (The expression of CD42b on the surface of platelets decreased significantly, while the expression of CD41 and CD61 was normal.)
Discussion
The abnormality of platelet glycoprotein (GP) Ib-IX-V complex is considered to be the foundation of BSS. GP Ib-IX-V complex has two important roles in platelet function: (1) It acts as a VWF receptor to mediate platelet adhesion to the injury vascular endothelium. It activates the integrin receptor αⅡbβ3 through the downstream signaling pathway to form stable platelet thrombus. (2) It maintains platelet structure integrity and cell morphology. The complex contains four distinct transmembrane proteins: GP Ibα, GP Ibβ,GP IX, and GP V [Citation6]. GP Ibα, GP Ibβ, and GP IX are essential for the efficient surface expression of the complex. A genetic mutation in the genes for any of these proteins may lead to quantitative or qualitative defects of the complex, affecting its binding to VWF factor and lead to failure of platelet adhesion to vascular endothelial connective tissue. To date, no GP V gene mutations have been reported, possibly because it is loosely bonded, and it appears that deletion of GP V has no effect on the expression and function of the complex [Citation7].
A literature review revealed considerable variability in the clinical manifestations and age of onset in patients with BSS. The clinical features of BSS generally include skin and mucous membrane bleeding, nosebleeds, gingival bleeding, and menorrhagia (female patients), or bleeding after surgery and trauma. Most patients presented with mild bleeding symptoms or only bleeding after surgery and trauma. Severe manifestations such as intracerebral hemorrhage and death because of bleeding are rarely reported [Citation8]. Regarding to the onset age, most patients are diagnosed with BSS in childhood due to a bleeding tendency since birth or since childhood. However, the diagnosis can be delayed until adulthood in milder forms of BSS because if the manifestations are not clinically significant or present later [Citation9].
Platelet count is variable in BSS, usually between 20 and 100 × 109/L, and the bleeding time ranges from mildly to severely prolonged [Citation7]. Clinically BSS can easily be misdiagnosed as ITP, and result in patients receiving unnecessary treatments such as glucocorticoids and splenectomy [Citation10, Citation11]. The difference between ITP and BSS is that BSS is associated with the presence of very large platelets. Noris et al. [Citation12] reported that an MPV threshold value of 12.4 fL distinguishes inherited macrothrombocytopenia from ITP with 91% specificity. Therefore, the blood cell counts and blood smear should be performed first in differential diagnosis. Moreover, automated hematology analyzers can mistake large platelets for lymphocytes; therefore, optical or manual counting are recommended for obtaining accurate platelet counts [Citation13]. In the platelet aggregation test, BSS platelets generally show a defective response to ristocetin-induced aggregation that cannot be corrected by adding normal plasma. This can be used to distinguish BSS from von Willebrand disease [Citation7, Citation14]. However, in this case, the ADP- and arachidonic acid-induced platelet aggregation were also inhibited, which is inconsistent with typical BSS. LTA is a classic method to detect platelet function, which has low cost, but is easily affected by various factors. The platelet counts were low at the time of testing, which may have contributed to lower platelet aggregation. We noted that the ristocetin-induced aggregation was the lowest, whereas the ADP- and arachidonic acid-induced platelet aggregation were closer to normal, therefore, we speculate that ristocetin-induced aggregation is the best indicator of actual platelet function.
Moderate or significant reduction of platelet glycoproteins (GP42b and GP42a) detected by flow cytometry is an important basis for diagnosing BSS. In addition, molecular techniques to identify genetic abnormalities are crucial to the diagnosis of BSS. In this case, ITP was suspected previously based on the decreased platelet count and two bone marrow cytology results. The outcome of further flow cytometry and gene test indicated the loss of expression of GPIb and three novel heterozygous mutations (c.95_101del, c.1012del and c.1616T>G) in GP1BA gene. The history of bleeding since childhood, large platelets, and low platelet aggregation function, made the diagnosis of BSS conclusive.
The gene mutations reported of BSS include mutations in GP1BA, GP1BB, and GPIX. In total, at least 114 mutations have been reported in BSS, of which 53 GP1BA gene mutations have been reported in 253 patients, 29 GP1BB gene mutations have been reported in 90 patients, and 32 GP9 gene mutations have been reported in 114 patients. A summary of the mutation sites reported in previous studies is provided in . Gene mutations are most frequently reported in the GP1BA gene (46.5%). The mode of inheritance in BSS is generally autosomal recessive, but rare autosomal dominant cases have been reported. Fewer than ten autosomal dominate variants of BSS have been reported, all of which were in the GP1BA gene. The clinical symptoms of autosomal dominate BSS are generally mild. In addition, BSS can be divided into mono-allelic BSS (mBSS) and bi-allelic BSS (bBSS), according to the number of gene mutations present. The clinical manifestations of mBSS are generally milder than those of bBSS, and are primarily due to the mild to moderate thrombocytopenia [Citation15]. The two novel heterozygous mutations (c.95_101del and c.1012del) confirmed as autosome recessive by the family pedigree in this case have not been reported previously. The two mutations of BSS caused mild symptoms and a low platelet count, which made it easy to misdiagnose as ITP. Therefore, gene testing is crucial to the early diagnosis of BSS and is recommended in patients with suspected BSS.
Table 1. Summary of mutation sites reported in previous studies.
In conclusion, we report a pediatric patient with a prolonged history of thrombocytopenia, who was finally diagnosed with BSS through genetic analysis. We identified two novelmutation sites in the GP1BA gene, c.95_101del and c.1012del, as the cause of BSS, enriching knowledge of the GP1BA mutation spectrum. This case highlights the importance of genetic testing in making the diagnosis of BSS.
Author contributions
Senlin Zhang, Jing Ling, and Kai Cui: study design, manuscript draft, and data collection. Shihong Zhan, Jiajia Zheng, and Wenyi Wang collected clinical data. Junjie Fan and Shaoyan Hu: data interpretation, analysis, and manuscript revision. All authors have read and approved the final manuscript.
Acknowledgments
We would like to thank Editage (www.editage.com) for the language editing provided for this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- López JA, Andrews RK, Afshar-Kharghan V, et al. Bernard–Soulier syndrome. Blood. 1998;91(12):4397–4418. doi:10.1182/blood.V91.12.4397
- Andrews RK, Berndt MC. Bernard–Soulier syndrome: an update. Semin Thromb Hemost. 2013;39(6):656–662. doi:10.1055/s-0033-1353390
- Berndt MC, Andrews RK. Bernard–Soulier syndrome. Haematologica. 2011;96(3):355–359. doi:10.3324/haematol.2010.039883
- Savoia A, Kunishima S, De Rocco D, et al. Spectrum of the mutations in Bernard–Soulier syndrome. Hum Mutat. 2014;35(9):1033–1045. doi:10.1002/humu.22607
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
- Bryckaert M, Rosa JP, Denis CV, et al. Of von Willebrand factor and platelets. Cell Mol Life Sci. 2015;72(2):307–326. doi:10.1007/s00018-014-1743-8
- Lanza F. Bernard–Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet J Rare Dis. 2006. doi:10.1186/1750-1172-1-46.
- Grainger JD, Thachil J, Will AM. How we treat the platelet glycoprotein defects: Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults. Br J Haematol. 2018;182(5):621–632. doi:10.1111/bjh.15409
- Alamelu J, Liesner R. Modern management of severe platelet function disorders. Br J Haematol. 2010;149(6):813–823. doi:10.1111/j.1365-2141.2010.08191.x
- Noda M, Fujimura K, Takafuta T, et al. Heterogeneous expression of glycoprotein Ib, IX and V in platelets from two patients with Bernard–Soulier syndrome caused by different genetic abnormalities. Thromb Haemost. 1995;74(6):1411–1415. doi:10.1055/s-0038-1649956
- LiC PD, Roth GJ. Bernard–Soulier syndrome with severe bleeding: absent platelet glycoprotein Ib alpha due to a homozygous one-base deletion. Thromb Haemost. 1996;76(5):670–674. doi:10.1055/s-0038-1650640
- Noris P, Klersy C, Gresele P, et al. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: a multicentric, real life study. Br J Haematol. 2013;162(1):112–119. doi:10.1111/bjh.12349
- Alamelu J, Liesner R. Modern management of severe platelet function disorders. Br J Haematol. 2010;149(6):813–823. doi:10.1111/j.1365-2141.2010.08191.x
- Pham A, Wang J. Bernard–Soulier syndrome: an inherited platelet disorder. Arch Pathol Lab Med. 2007;131(12):1834–1836. doi:10.5858/2007-131-1834-BSAIPD
- Leinøe E, Brøns N, Rasmussen AØ, et al. The Copenhagen founder variant GP1BA c.58T > G is the most frequent cause of inherited thrombocytopenia in Denmark. J Thromb Haemost. 2021;19(11):2884–2892. doi:10.1111/jth.15479