
Abstract
Objectives: To evaluate the characteristics and outcomes of treatment of patients with pheochromocytoma at Inkosi Albert Luthuli Central Hospital (ILACH) in Durban, South Africa over 14 years.
Design: Retrospective chart review.
Setting and subjects: Patients with pheochromocytoma attending the endocrinology clinic at IALCH between 2012 and 2016 were studied.
Outcome measures: Clinical, biochemical and radiological data were collected at presentation, on discharge, one year and five years after surgical intervention; tumour characteristics, histopathological features and surgical outcome were also assessed.
Results: The analysis included 35 patients (mean age 33.2 ± 15.7 years; 60% female).
Headache (68.6%), palpitation (60%) and sweating (57.6%) were the three most common presenting symptoms; hypertension was the predominant clinical finding (85.7%). Most pheochromocytomas were sporadic (82.9%), adrenal gland tumours (68.6%) and benign (77.1%); of eight patients with malignant tumours, two were familial. Adrenalectomy was undertaken in the majority (n = 34; 97.1%); 55.2% were large tumours. The use of adjunctive radiotherapy (n = 4; 11.4%) and chemotherapy (n = 1; 2.9%) was low. There was low overall mortality (5.7%), but 57.6% developed intraoperative hypotension. At one year postoperatively, 80% (n = 28) of patients were defined as cure, biochemically in 23 (82.1%) and with radiology in five (17.9%).
Conclusions: Most patients presenting to IALCH had large intra-abdominal tumours with high cure rate, low mortality but a high rate of perioperative complications. Late presentation and large tumour size was a feature.
Introduction
Pheochromocytomas are rare neuroendocrine tumours with variable clinical presentation including hypertension.Citation1−4 These are benign or malignant catecholamine-producing tumours that arise from neural crest chromaffin cells, which are located in the adrenal medulla or extra- adrenal paraganglia. The majority are those arising from the adrenal medulla and are called pheochromocytomas (80–85%) while tumours arising outside the adrenal gland are referred to as extra-adrenal pheochromocytomas or catecholamine-secreting paragangliomas (15–20%). The only criterion for distinguishing malignant pheochromocytoma from benign lesions is the presence of metastases of chromaffin tissue at sites not usually present.Citation1,2,4−10
Although it can be cured with surgery, when undiagnosed pheochromocytoma can be fatal.1 Therefore, early detection of pheochromocytoma is crucial to prevent the associated life-threatening cardiovascular complications (cardiomyopathy, myocardial infarction, cardiac arrhythmia and stroke).Citation1,2,6 The reported prevalence is 0.1–0.6% in hypertensive patients with an annual incidence of 2–8 cases per million.Citation1,2 , Citation11,12 Age at presentation is typically in the third–fifth decades of life with a female preponderance.Citation1,3
While most pheochromocytomas are sporadic, at least 30% of these tumours may be hereditary. The inherited familial syndromes associated with pheochromocytoma include multiple endocrine syndrome type 2 (MEN 2), von Hippel-Lindau (VHL) syndrome, neurofibromatosis type 1 (NF-1), and familial paraganglioma.Citation1,2,10,11
The clinical features of pheochromocytomas are mainly due to the production of catecholamines, with the commonest symptoms being headache, palpitation and diaphoresis. However, because of the rarity of these tumours, it is not cost-effective to screen all hypertensive patients; the recommendation is that only patients with a high index of suspicion, namely those with features of hyper-adrenergic spells, a familial syndrome and patients with adrenal incidentaloma be investigated.Citation1,2,6,11,12
When the tumour is suspected clinically, biochemical confirmation (plasma and urinary catecholamines (adrenaline, noradrenaline)) and their metabolites (metanephrine and normetanephrine) is required followed by tumour localisation using various imaging studies.Citation1 − Citation3,5,10−13 It is advised that plasma tests should be confined to patients with a high clinical suspicion to avoid a high rate of false-positive results.Citation16 When compared with urinary-fractionated metanephrines, which have high sensitivity and specificity (91–98%), plasma-fractionated metanephrines have higher sensitivity (96–100%) but lower specificity (85–89%).Citation1,2,11,17
Surgical resection followed by postoperative monitoring is the safest and the most effective therapeutic intervention for pheochromocytoma.Citation1,2,4,9,11,14,18
Although there are several reports on pheochromocytoma from centres in developed countries,Citation19−22 there is limited information available from developing regions of the world such as sub-Saharan Africa. This is probably related to limited healthcare resources and laboratory facilities for the diagnosis of pheochromocytoma in Africa. Available reports from South Africa are limited to studies reported from a tertiary referral centre in Johannesburg. In 54 patients who presented with pheochromocytoma between 1980 and 2009, there was low mortality (7.4%; n = 4); > 90% of those patients who were diagnosed based on biochemical profile before the histopathological confirmation had a good outcome.Citation23
To date there are no reports of the clinical presentation and outcome of patients with pheochromocytoma in Durban. This study was undertaken to determine the characteristics and outcome of pheochromocytoma patients who attended and were managed in the Endocrinology Clinic at Inkosi Albert Luthuli Central Hospital (IALCH) in Durban, South Africa.
Study design and methods
Study design
A retrospective chart review was undertaken on all patients with pheochromocytoma who were managed at the Endocrinology Clinic at Inkosi Albert Luthuli Central Hospital (IALCH) in Durban, South Africa between September 1, 2002 and December 31, 2016. IALCH is a public tertiary referral teaching hospital with a total bed capacity of almost 900; this hospital provides specialist and subspecialist healthcare services for more than 10 million inhabitants in the province of KwaZulu-Natal and parts of the Eastern Cape Province. The study was approved by the Biomedical Research Ethics Committee of the University of KwaZulu-Natal (reference: BE307/13).
Study methods
Clinical information on all patients with pheochromocytoma was retrieved from hospital records; the hospital has a computerised data management system, which allows for record keeping and retrieval. For each patient, information at presentation, post-surgery (at discharge, one year, five years and > 10 years, where applicable) was recorded. These included demographic details, clinical presentation, associated co-morbidities, physical examination, biochemical and genetic testing (where applicable), radiological, surgical and histological findings. No patients were excluded from the study over the study period.
Laboratory tests were undertaken by the Chemical Pathology Laboratory at IALCH or its referral laboratory. Pre- and post-surgery hormonal assessment for pheochromocytoma: 24 h urine catecholamines (adrenaline, noradrenaline and dopamine) were available for all patients; serum chromogranin A has been measured routinely since 2009 and screening for MEN commenced routinely in 2007. Other routine biochemical and haematological tests (plasma glucose, urea and electrolytes, serum calcium, full blood count) were also available.
The biochemical criteria used for the diagnosis of pheochromocytoma was a 24 h urine catecholamine level > 2 times the upper limit of the normal reference range for the IALCH laboratory.Citation2,16 The normal reference ranges for urine catecholamines at the IALCH laboratory are as follows: 24 h urine adrenaline 0–86 nmol/24 h; noradrenaline 0–615 nmol/24 h; dopamine 0–3786 nmol/24 h. The reference range for serum chromogranin A is 0–101.9 ng/ml. Urine catecholamines were measured with reverse phase high-pressure liquid chromatography (HPLC) (Recipe kit, Recipe, Munich, Germany); serum chromogranin A was measured with an automated immunofluorescent method (Thermo Fischer kit, Thermo Fischer Scientific, Henningsdorf, Germany).
Screening for MEN 2A included measurement of serum parathyroid hormone, calcitonin, 24 h urine free cortisol and thyroid ultrasound. Molecular genetic testing for other familial disorders was also done at the time of the diagnosis, where these were considered. In addition, serum calcium and random blood glucose levels were done for all patients preoperatively.
Radiological investigations were performed to determine tumour location (whether adrenal or extra-adrenal and unilateral or bilateral lesion), tumour size, evidence of metastasis and associated calcification, necrosis and or haemorrhage. These included ultrasonography, computerided tomography (CT) scan, magnetic resonance imaging (MRI), I123metaiodobenzylguanidine scintigraphy (MIBG) and positron emission tomogram (PET) scan. Malignant pheochromocytoma was considered if lesions were found in other organs. Most patients had preoperative and multiple postoperative CT scans. Postoperatively all the CT reports were reviewed for the presence of residual tumour and/or evidence of local or metastatic tumour recurrence.
Cure was defined biochemically in secretory tumours and these include all tumours in whom the preoperative levels were in the diagnostic range (> 2 x ULN) as well as any elevation above the normal range. In this study, only normal 24 h urine catecholamines were used to define biochemical cure; chromogranin A was not used as a biochemical criterion as it was not routinely measured until 2009 and because of lack of concordance with urine catecholamines in cured patients. In patients with non-secretory (non-functional) tumours, cure was determined radiologically by absence of tumour on radiological imaging post-surgery.
Follow-up was undertaken by the Endocrinology Clinic for all patients and included clinical examination, repeat imaging and hormonal testing.
Statistical analysis
Statistical analysis was performed using STATA† (V13.1; STATA/IC, College Station, TX, USA). Descriptive statistics were used for demographic and clinical data. Frequencies and percentages were used for categorical data. As numerical data did not meet the assumption of normality using the Shapiro–Wilk test, medians and interquartile ranges (IQR) were reported. Subgroup comparisons for categorical variables were compared with Fisher’s exact test due to the small cell sizes and the two-sample Wilcoxon rank-sum (Mann–Whitney) test was used for continuous variables. A p-value < 0.05 was considered statistically significant.
Results
Patient characteristics based on age, gender and race
A total of 35 patients (M:F; 14:21), with suspected pheochromocytoma were reviewed between 2002 and 2016. Mean age at presentation was 33.2 ± 15.7 years (age range 11–69 years). Seven patients presented during childhood (< 18 years). In all patients, the diagnosis of pheochromocytoma was made ante-mortem. The majority (60%; n = 21) were African (black) and 28.6% (n = 10) were Asian Indians; Whites (8.6%; n = 3) and mixed-race (Coloured) (2.9%; n = 1) patients constituted the remainder (Table ).
Table 1: Characteristics of patients with pheochromocytoma at presentation (n = 35)
Presenting clinical features
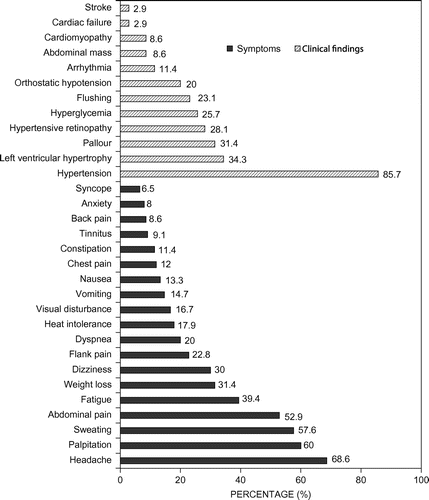
Figure shows the clinical features at presentation. The three most common symptoms were headache (68.6%), palpitation (60%), and sweating (57.6%). Abdominal pain, fatigue and weight loss were also frequent symptoms. The majority of patients (n = 30; 85.7%) presented with hypertension; of these, 65.7% had sustained hypertension and 20% had paroxysmal hypertension. Clinical features of established hypertension, namely retinopathy (grade I (n = 4; 12.5%), grade II (n = 4; 12.5%) and grade III (n = 1; 3.1%)) and left ventricular hypertrophy were found in 28.1% and 34.3%, respectively. Orthostatic hypotension was a feature in seven (20%) patients. Three patients were pregnant at presentation; one in the second trimester and two in the third trimester.
Figure 1: Presenting clinical features in patients with pheochromocytoma (n = 35). ■ represents presenting symptoms and □ represents clinical findings.
Co-morbidities at presentation included diabetes mellitus (n = 3; 8.6%) and chronic kidney disease (n = 2; 5.7%).
Tumour location and characteristics
All the patients presented with intra-abdominal tumours (Table ). Of these, 24 (68.6%) originated from the adrenal gland, 10 (28.6%) were located extra-adrenally and one (2.9%) was a combined adrenal and extra-adrenal tumour. Of the 25 adrenal tumours, the majority (88%; n = 22) were unilateral and right-sided (68.2%; n = 15) tumours; three (12%) were bilateral adrenal tumours. Of the 11 extra-adrenal tumours, three were located in the urinary bladder, seven in the lower abdomen and one in the Organ of Zuckerkandl.
Table 2: Classification of pheochromocytoma in patients with pheochromocytoma (n = 35)
Malignancy was diagnosed in eight (22.9%) patients (four adrenal; three extra-adrenal; one combined adrenal and extra-adrenal) and 27 (77.1%) were considered benign tumours. Lymph node was the most common site of metastasis accounting for 62.5% (n = 5) of all malignant tumours; pulmonary and vascular (inferior vena cava and unspecified blood vessel invasion) metastasis was found in 25% (n = 2).
The majority (n = 29; 82.9%) of patients had sporadic tumours. Only six (17.1%) patients had familial syndromes; of these, five had VHL (four of them from the same family: father, son and two daughters) and one had MEN type 2a based on the concomitant presence of biochemical (raised calcitonin level) and radiological features of medullary thyroid carcinoma on ultrasound and CT scan. Furthermore, five of the familial cases had adrenal tumours (one bilateral and four unilateral) and only one case was documented as extra-adrenal.
Of the 35 patients, 20 (57.1%) were diagnosed on the basis of biochemical results (i.e. > 2 times ULN of 24 h urine catecholamine) and the diagnosis in the remainder (n = 15; 42.9%) was incidental (either clinical and/or radiological) as they were non-functional (non-secretory) tumours or had mild elevation in 24 h urine catecholamine (i.e. less than twice ULN). Secretory tumours accounted for 77.1% (n = 27). Noradrenaline was the predominant (92.6%) catecholamine secreted; adrenaline (37%) and dopamine (11.1%) constituted the remainder.
Biochemical characteristics
In 34 patients in whom it was measured, median (IQR) 24 h urine catecholamine (nmol/24hr) was as follows: adrenaline 31 (12–142), noradrenaline 1445 (536–3384), dopamine 1319 (500–2252); mean 24 h urine adrenaline (144.17 ± 268.20) was 1.7 times and noradrenaline (3064.70 ± 3727.20) five times above the reference range. In the 25 patients in whom they were measured, median (IQR) serum chromogranin A was 739.4 (218.5–1399) ng/ml; the mean value (1173.12 ± 1466.37) was 11.5 times above the reference range. Mean serum calcium was 2.40 ± 0.16 mmol/l and 23% (n = 8) had hypercalcemia. Median (IQR) for plasma glucose was 5.7 mmol/l (4.6–7.9); hyperglycaemia was found in 25.7% (n = 9) of these, three were diagnosed with diabetes mellitus prior to presentation.
Radiological tests
Radiological characteristics are given in Table . At presentation, the majority (n = 31; 88.6%) had CT scan for initial tumour localisation. Four patients (11.4%) had MRI scans (three subjects were pregnant and the indication for the MRI in the fourth patient was unclear); of these, one showed evidence of metastasis. In all (100%), CT or MRI (where applicable) scans correctly identified tumours before surgery. Baseline MIBG scans were done in 60% (n = 21); of these, 19 (90%) correctly identified the tumour preoperatively; two tumours that were not identified by MIBG scan were non-secretory. PET scans were performed in 25.7% (n = 9) for identification of distant metastasis where results of CT or MIBG scans were inconclusive. Tumour necrosis and associated calcification or haemorrhage was recorded radiologically in five (14.3%) patients (four on CT scan and one on MIBG study).
Table 3: Radiological characteristics in patients with pheochromocytoma at presentation (n = 35)
Treatment modalities
Table outlines the mode of therapy. The majority (n = 34; 97.1%) of patients had adrenal surgery as primary therapy. Anterior midline adrenalectomy was the most common surgical approach (n = 19; 54.3%) and laparoscopic adrenalectomy (n = 15; 42.9%) in the remainder.
Table 4: Therapeutic modalities and perioperative complications in patients with pheochromocytoma (n = 35)
Medical therapy (alpha-adrenergic blockers) alone was used only in one patient who refused surgery.
Other therapeutic modalities included MIBG-based radiotherapy for four (11.4%) patients (one each for massive tumour extension to IVC, distant metastasis to the lung, residual metastatic tumour and having local recurrent disease); chemotherapy was utilised in one (2.9%) subject who had metastatic disease.
Alpha-adrenergic blockers were the commonest preoperative anti-hypertensive medication used (n = 31; 88.6%), followed by calcium channel (n = 18; 51.4%) and beta-adrenergic blockers (n = 17; 48.6%).
Perioperative complications are presented in Table . Two patients (5.9%) died: one intraoperative with acute haemorrhagic shock and one on day four postoperatively, due to septic shock. Of the 34 patients who were treated surgically, 22 (64.7%) experienced a complication. The most frequently documented intraoperative complication was hypotension (55.9%), followed by intraoperative haemorrhage (20.6%). At surgery, tumour size was recorded in most cases (n = 29; 82.9%) with all malignant and 21 benign tumours; the largest dimensions were recorded as ranging from 3 cm to 12 cm (with mean 6.4 cm). Malignant tumours (mean size 9 cm) were larger than benign (mean size 5.6 cm) tumours. Large tumour size (> 6 cm) was found in 55.2% (n = 16).
Tumour histology
All tumours were biopsied and histology confirmed adrenal pheochromocytoma in 68.6% (n = 24) and paraganglioma in 28.6% (n = 10); histological features of both adrenal and extra-adrenal tumours were documented in one patient (2.9%). Small nests (zellballen) of chromaffin-staining cells were the main histological manifestation of adrenal and extra-adrenal pheochromocytoma. Malignancy was diagnosed based on the presence of metastatic chromaffin tissue in the affected organs, namely lymph nodes (n = 5) and lung (n = 2) or by radiological documentation (CT/MRI/MIBG) of the metastasis (n = 1) to the liver and bone.
Patient outcome on discharge, one year and five years post-surgery
Of the 35 patients in whom pheochromocytoma was confirmed histologically, 80% (n = 28) were defined as cured one year postoperatively. Of these, 23 (82.1%) had secretory tumours and deemed as cure by the biochemical definition; five (17.9%) patients with non-secretory tumours were deemed as cure radiologically. On the other hand, seven (20%) patients were deemed ‘not cured’; these included two patients at one year postoperatively, two at five years postoperatively, one who refused surgery and two who died within a week of surgery.
No significant difference was found for baseline biochemical and radiological features between cured and non-cured pheochromocytoma patients (Table ).
Table 5: Baseline biochemistry in cured (n = 28) and not-cured (n = 7) patients with pheochromocytoma⃰
Discussion
This study on characteristics and outcomes of treated patients with pheochromocytoma has shown that all tumours presented intra-abdominally, the majority located in the adrenal gland and most were benign tumours. Cure was documented in the majority and, despite the high rate of perioperative complications, the overall mortality was low. Clinical presentation in most patients included the classical pheochromocytoma triad associated with hypertension.
The pheochromocytoma triad (headache, 68.6%; palpitations, 60%; and sweating, 57.6%) were the main presenting symptoms in the majority of patients, confirming the classical hyper-adrenergic spells of the tumours. This is in keeping with the available literature, which reports headaches in 55–90%, palpitations in 50–77% and sweating in 40–74% of patients with pheochromocytoma.Citation2,3,6,23
Hypertension was the most frequent presenting clinical finding, found in 85.7% in the current study, which is quite close to the figures (90–100%) from most available reports,Citation2,3,6,23 except one study that reported a low rate (45%).Citation19 Furthermore, in our study, sustained hypertension (65.7%) was more common than paroxysmal hypertension (20%), similar to studies from Asia and Europe,Citation9,19,22 but higher than other studies, which report similar proportions for both types.Citation3,6,23 Hypertension persisted in 33.3% after successful resection of the tumour (as defined biochemically and radiologically); similar findings have been reported from centres in AsiaCitation9,19 and Sweden,Citation22 with rates of 10–35%. This may be related to the late presentation with target organ (renal) involvement from hypertension or the coexistence of essential hypertension in this study.
Pheochromocytoma in pregnancy can cause death for both mother and foetus with high mortality rate that can reach 50% in both.Citation24 Only three patients (8.6%) in this study were diagnosed during pregnancy. However, as in the larger series from Johannesburg,Citation23 there was no maternal mortality in this study and this is probably accounted for by the diagnosis being made ante-partum and institution of appropriate treatment.
Adrenal pheochromocytoma accounted for the majority in this study (68.6%). Available reports are variable with some studies reporting high rates (78–92%)Citation1,3,21,22,25 and only one study reporting a lower rate (61%) of adrenal tumours.Citation23
Consistent with the available literature,Citation3,22 benign tumours (77.1%) were far more common than malignant in our setting; however, the frequency of malignant pheochromocytoma (22.9%) is higher than the 4–15% reported;Citation15,20,23 and probably accounts for the high proportion of large tumours found in this study.
As in other studies,Citation3,14,23 familial syndromes were diagnosed in the minority (n = 6; 17.1%). In our setting the frequency of patients with MEN2A is higher than that reported from Johannesburg (1.9%)Citation23 but lower than that reported by Safwat et al. (14.9%)Citation,3 Lo et al. (10.4%)Citation19 and Fernández-Calvet et al. (7.1%).Citation22 Unlike other studies where genetic tests were used to diagnose inherited cases,Citation5,23 in this study familial tumours were diagnosed primarily on the basis of the positive family history of pheochromocytoma and presence of biochemical and radiological features of other components of inherited pheochromocytoma syndromes such as bilateral tumours, renal cell carcinoma, or medullary thyroid carcinoma. Selective genetic testing has only recently become available at our centre; only two of the six familial pheochromocytoma patients underwent genetic testing and no gene mutation was detected.
In the current study, 57.1% of patients were diagnosed based on biochemical results, much lower than that reported (88.8–96.5%) in other studies,Citation3,6 and is probably accounted for by the low sensitivity and specificity of the tests used.
Similar to other studies,Citation3,19,25 anatomical imaging (CT or MIBG scans) was able to correctly localise all tumours preoperatively. MIBG correctly identified the tumour in 90%; this is higher than that reported from a Chinese study (79%),Citation19 but similar to other studies (88–98%),Citation26–28 and may be related to large tumour sizes in this study.
An anterior midline surgical approach was used in just over half our patients (54.3%), in view of the fact that large tumours were identified with preoperative localisation. This is compatible with the study by Lo et al. Citation19 (mean tumour size 6.4 cm), but higher than in other studies (mean tumour size 4.8 cm).Citation9,29,30
Tumour size in our study (6.4 cm) was larger than that recorded in other studies (mean 5.2 cm).Citation3,4,7,8 This may be accounted for by the late presentation or delayed referral for specialised care. However, in concordance with other studies,Citation19,25 malignant tumours (mean size 9 cm) were larger than benign (mean size 5.6 cm).
In the present study, for patients with metastatic tumours, radiotherapy (11.4%) and chemotherapy (2.9%) were both used as adjunctive therapies and this is in keeping with the 10–21% reported for radiotherapy use,Citation31,32 but lower than that for use of chemotherapy (16–30%).Citation31,32
In the current study, 80% were deemed as cure (biochemically and/or radiologically) and is similar to results reported in other studies (79–92.6%).Citation2,15 , Citation19,23
Available information on mortality rates is variable. The mortality of 5.9% found in this series is lower than that reported by Modigliani et al. (13%)Citation20 whose results may be related to the high proportion of familial cases of pheochromocytoma (all patients had either MEN I2A or MEN I2B) in that study. By contrast, low mortality rates have also been reported in the Johannesburg study (7.4%).Citation23
This study has several limitations including that it is a retrospective chart review in a single centre, the small number of patients and the biochemical tests used for diagnosis. However, the last report from this country was in 2011 from Johannesburg and this is the first from this centre, and may serve to highlight the late presentation and the need for a high index of suspicion at a primary health care level to facilitate early referral.
Conclusion
Patients presenting to IALCH had intra-abdominal tumours with high cure rates despite large tumour size. Mortality was low but there was a high rate of perioperative complications. The clinical presentation and large tumours probably relate to late presentation. A high index of suspicion at primary health care levels and a multidisciplinary approach is necessary to treat these rare tumours effectively. Large prospective studies are required to confirm the results of this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Declaration on copyright and originality of paper
We confirm that the work is original and has not been published elsewhere, nor it is currently under consideration for publication elsewhere.
Declaration regarding authorship
We have the right to publish the paper.
Ethics committee approval
Ethics committee (Biomedical Research Ethics Committee of the University of KwaZulu-Natal) approval has been obtained for original study and is clearly stated in the methodology. (Reference: BE307/13).
References
- Young WF . Adrenal causes of hypertension: pheochromocytoma and primary aldosteronism. Rev Endocr Metab Disord. 2007;8(4):309–20.10.1007/s11154-007-9055-z
- Lenders JWM , Eisenhofer G , Mannelli M , et al. Lancet. 2005;366:665–75.10.1016/S0140-6736(05)67139-5
- Safwat AS , Bissada NK , Seyam RM , et al. The clinical spectrum of pheochromocytoma: analysis of 115 patients. BJU Int. 2008;101(12):1561–4.10.1111/j.1464-410X.2008.07430.x
- Plouin PF , Amar L , Dekkers OM , et al . European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a pheochromocytoma or a paraganglioma. European Journal of Endocrinology. 2016;174(5):G1–G10.10.1530/EJE-16-0033
- Cano Megías MC , Rodriguez Puyol DR , Fernández Rodríguez LF , Sención Martinez GLS , Martínez Miguel PM . Pheochromocytoma -paraganglioma: Biochemical and genetic diagnosis. Nefrología. 2016;36(5):481–8.10.1016/j.nefro.2016.03.010
- Mannelli M , Lenders JWM , Pacak K , Parenti G , Eisenhofer G . Subclinical pheochromocytoma. Best Practice & Research Clinical Endocrinology & Metabolism. 2012;26(4):507–15.10.1016/j.beem.2011.10.008
- Kulkarni MM , Khandeparkar SGS , Deshmukh SD , et al . Risk Stratification in Paragangliomas with PASS (Pheochromocytoma of the Adrenal Gland Scaled Score) and Immunohistochemical Markers. Journal of Clinical and Diagnostic Research. 2016;10(9):1–4.
- Garrahy A , Casey R , Wall D , Bell M , O’Shea PM . A review of the management of positive biochemical screening for pheochromocytoma and paraganglioma: a salutary tale. Int J Clin Pract. 2015;69(7):802–9.10.1111/ijcp.12612
- Rao N , Ramachandran R , Tandon N , Singh P , Kumar R . Laparoscopic adrenalectomy for pheochromocytoma-does size matter? A single surgeon comparative study. Translational Andrology and Urology. 2016;5(5):780–3.10.21037/tau
- Donckier JE , Michel L . Pheochromocytoma : state-of-the-art. Acta Chirurgica Belgica. 2010;110(2):140–8.10.1080/00015458.2010.11680587
- Estey MP , Diamandis EP , Eisenhofer G , et al . Pheochromocytoma. Clinical Chemistry. 2013;59(3):466–72.10.1373/clinchem.2012.182246
- van Berkel A , Lenders JWM , Timmers HJLM . Diagnosis of endocrine disease: biochemical diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol. 2014;170(3):R109–R119.10.1530/EJE-13-0882
- Eisenhofer G , Tischler AS , de Krijger RR . Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: from routine laboratory methods to disease stratification. Endocr Pathol. 2012;23(1):4–14.10.1007/s12022-011-9188-1
- Chen H , Sippel RS , O’Dorisio MS , et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775–83.10.1097/MPA.0b013e3181ebb4f0
- Plouin PF , Amar L , Lepoutre C . Pheochromocytomas and functional paragangliomas: clinical management. Best Pract Res Clin Endocrinol Metab. 2010;24(6):933–41.10.1016/j.beem.2010.10.002
- Boot C , Toole B , Johnson SJ , Ball S , Neely D . Single-centre study of the diagnostic performance of plasma metanephrines with seated sampling for the diagnosis of pheochromocytoma /paraganglioma. Ann Clin Biochem. 2017;54(1):143–8.10.1177/0004563216650463
- Young WF Jr . The incidentally discovered adrenal mass. N Engl J Med. 2007;356(6):601–10.10.1056/NEJMcp065470
- Munakomi S , Rajbanshi S , Adhikary S . Case report: agiant but silent adrenal pheochromocytoma - a rare entity [version 1; referees: 2 approved]. F1000Research. 2016;5:290.
- Lo CY , Lam KY , Wat MS , et al. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212–5.10.1016/S0002-9610(00)00296-8
- Modigliani E , Vasen HM , Raue K , et al . Pheochromocytoma in multiple endocrine neoplasia type 2: European study. J Intern Med. 1995;238(4):363–7.10.1111/joim.1995.238.issue-4
- Stenstro G , Svardsudd K . Pheochromocytoma in Sweden 1958– 1981. JIM. 1986;220(3):225–32.
- Fernández-Calvet L , García-Mayor RVG . Incidence of pheochromocytoma in South Galicia, Spain. J Intern Med. 1994;236(6):675–7.10.1111/joim.1994.236.issue-6
- Huddle KRL . Pheochromocytoma in black South Africans - a 30-year audit. S Afr Med J. 2011;101(3):184–8.10.7196/SAMJ.4320
- Schenker JG , Granat M . Pheochromocytoma and pregnancy — an updated appraisal. Aust NZJ Obstet Gynaec. 1982;22(1):1–10.10.1111/ajo.1982.22.issue-1
- Park J , Song C , Park M , et al . Predictive characteristics of malignant pheochromocytoma. Korean J Urol. 2011;52(4):241–46.10.4111/kju.2011.52.4.241
- Berglund AS , Hulthen UL , Manhem P , et al. Metaiodobenzylguanidine (MIBG) scintigraphy and computed tomography (CT) in clinical practice. Priamary and secondary evaluation for localization of pheochromocytomas. J Intern Med. 2001;249(3):247–51.10.1046/j.1365-2796.2001.00792.x
- Shapiro B . Imaging of catecholamine-secretion tumours: uses of MIBG in diagnosis and treatment. Baillieres Clin Endocrinol Metab. 1993;7(2):491–507.10.1016/S0950-351X(05)80185-5
- Van Der Horst-Schirvers AN , Jager PL , Boezen HM , et al. Iodine-123 metaiodobenzylguanidine scintigraphy in localising pheochromocytoma -experience and meta-analysis. Anticancer Res. 2006;26(2):1599–604.
- Shubin W , Weiyun C , Le S , et al. Risk factors for prolonged hypotension in patients with pheochromocytoma undergoing laparoscopic adrenalectomy: a single- center retrospective study. Sci Rep. 2017;7(1):5897.
- Carter YM , Mazeh H , Sippel RS , et al. Safety and feasibility of laparoscopic resection for large (≥6 cm) pheochromocytomas without suspected malignancy. Endocr Pract. 2012;18(5):720–6.10.4158/EP12014.OR
- Gedik GK , Hoefnagel CA , Bais E , et al. 131I-MIBG therapy in metastatic pheochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2008;35(4):725–33.10.1007/s00259-007-0652-6
- Huang KH , Chung SD , Chen SC , et al . Clinical and pathological data of 10 malignant pheochromocytomas: Long-term follow up in a single institute. Int J Urol 2007;14(3):181–185.10.1111/iju.2007.14.issue-3