
Abstract
Insulinomas are rare functional neuroendocrine tumours. These tumours present with symptoms of hypoglycaemia, adrenergic and neuroglycopenic symptoms. Insulinomas are usually small in size, making them difficult to locate. Diagnosis is often delayed in these cases resulting in lasting neurological complications, often not reversible after surgical removal of the insulinoma. We report a case of a 33-year-old HIV-positive female with delayed presentation of an insulinoma. She presented with a long history of neurological symptoms with lasting neurological sequelae and poor functional status, during which time the diagnosis of insulinoma was repeatedly missed.
Introduction
Insulinoma is one of the functional pancreatic tumours derived from the pancreatic ductal/acinar system that is rare but often missed.Citation1 Patients typically present with symptoms of unprovoked hypoglycaemia but as these symptoms are non-specific diagnosis is often delayed due to late presentation of patients or failure of healthcare workers to recognise the disease early. The main clinical features include adrenergic symptoms such as sweating and palpitations and neuroglycopenic symptoms including confusion, anxiety, stupor, convulsions and coma. The neuroglycopenic symptoms are often misdiagnosed as primary neurological disorders, which often causes a delay in diagnosis and treatment that can result in permanent neurological damage. Insulinomas are usually small in size and benign in nature. The tumours are malignant in less than 10% of cases.Citation2 Insulinomas are commonly sporadic in nature with only a few cases described as being associated with genetic mutations.Citation2 Localisation of these tumours presents a challenge to most clinicians as the standard techniques often used for localisation have poor sensitivity and specificity. Here we present a case of a patient who presented with recurrent hypoglycaemia complicated by neurological sequelae.
Case report
History
A 33-year-old female was referred to the Charlotte Maxeke Johannesburg Academic hospital from a care centre for assessment of her cognitive dysfunction and limb weakness. On arrival at the emergency room she was documented to have severe hypoglycaemia with a laboratory glucose of 1.3 mmol/l. She was not able to provide a history due to her poor cognitive function, but her family reported a one-year history of recurrent admissions to hospitals for hypoglycaemia. When given a glucose infusion she had clinical improvement post-incident and did not have further investigation done to ascertain the cause. She also had a history of difficulty walking progressively worsening associated with difficulty speaking, but no urinary or faecal incontinence and no sensory symptoms. She did not have a history of seizures.
On physical examination, she had frontal release signs, ataxic speech, wasting of upper and lower limbs with clawing of both hands and feet with generalised hypotonia and hyporeflexia. Her abdominal examination was normal.
Laboratory investigation () confirmed hypoglycaemia with a glucose of 1.3 mmol and with non-suppressed insulin of 23.8 mIU/l and C-peptide of 3 mg/l measured at the same time. Her serum cortisol level was 778 nmol/l excluding hypocortisolaemia as a cause for hypoglycaemia. Her thyroid function tests were normal as was her renal function (urea 2.3 mmol/l and creatinine of 53 µmol/l). Chromogranin A level was elevated with a level of 139.50 ng/ml (normal reference range less than 101.9 ng/ml). Urine for sulfonylurea screen was unavailable for testing. She also had mild normocytic anaemia with a haemoglobin level of 11.3 g/l. Her CD4 count was 549 with a viral load that was suppressed on highly active antiretroviral therapy for a duration of 6 years. All other blood results were within normal reference limits. In view of the history and findings the clinical impression was that of an insulinoma.
Table 1: Laboratory studies
To localise the insulinoma, an ultrasound of the abdomen was done which did not find any masses. An abdominal contrast-enhanced computer tomography scan was performed which showed a normal pancreas morphology with homogenous enhancement on all phases and no focal lesion.68 Ga-DOTA-octreotate PET/CT was performed, which showed avid active disease in the uncinated process of the pancreas with no evidence of nodal disease or distant metastasis ( and ). Endoscopic ultrasound was also performed, which revealed a well-visualised homogenously echoic pancreas with a well-circumscribed lesion with poor vascularity visualised in the uncinated process measuring 1.5 by 1.4 cm. There was no evidence of local lymph node involvement. Fine-needle aspiration was performed on the lesion. Cytology revealed sheets of benign epithelial cells, occasional pancreatic acinar cells and an isolated group of bland cells with neuroendocrine features in keeping with a neuroendocrine tumour.
Figure 1: Ga-DOTA octreotate PET/CT showing avid active disease in the uncinated process of the pancreas.
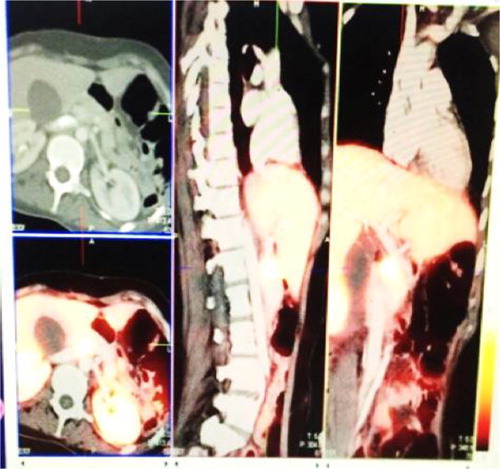
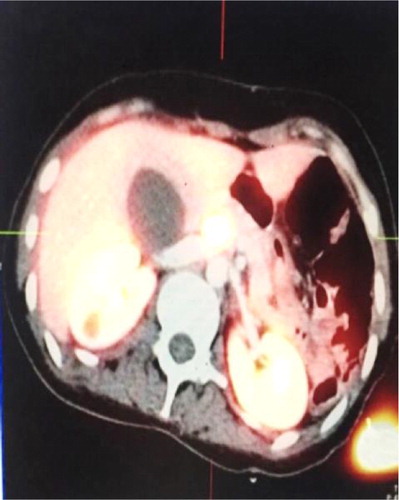
Figure 2: Enlarged Ga-DOTA octreotate PET/CT showing avid active disease in the pancreas.
Nerve conduction studies were also done, which indicated decreased amplitude and increased latency suggestive of an axonal neuropathy with secondary denervation.
Discussion
Insulinomas are rare functioning neuroendocrine tumours. They occur in 1–4 per million per year.Citation2 They are the most common class of neuroendocrine tumours. They occur more commonly in females, with an average age at presentation of 50 years.Citation3 Insulinomas are commonly sporadic in nature. Less than 10% are genetic in origin, associated with multiple endocrine neoplasia type 1 (MEN-1).Citation3 MEN-1 is an autosomal-dominant syndrome affecting mainly the parathyroid glands, anterior pituitary, endocrine pancreas and duodenum, due to inactivation of the MEN1 gene on chromosome 11q13. Of the patients with insulinoma associated with MEN1, the presentation is earlier and female predominant 2. The majority of patients with insulinomas present with hypoglycaemia in the fasting state although it can also occur in the post-prandial state.Citation3
Clinical presentation is due to hypoglycaemia, with adrenergic symptoms and neuroglycopenic symptoms. Adrenergic symptoms include palpitations, sweating and tremors. The neuroglycopenic presentation varies per case ranging from confusion to vague symptoms of weakness and objective hemiparesis/hemiplegia with symptoms dependent on the severity of the hypoglycaemia.Citation4
Neuroglycopenia as the main presenting complaint for hypoglycaemia is uncommon, only described in case reports. However, on careful questioning they are present in more than 90% of patients presenting with an insulinoma.Citation5 As symptoms are non-specific there is often a delay in diagnosis with an average time of one year from onset of symptoms to final diagnosis with patients often misdiagnosed with neurological or psychiatric disorders.Citation5
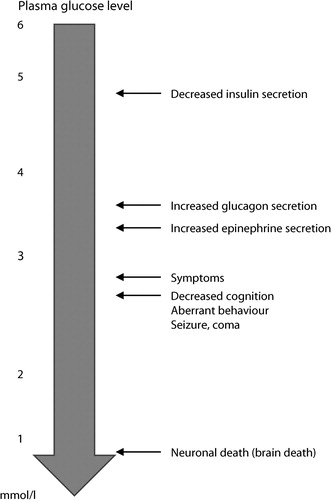
It is well known that glucose is an obligate supplier of fuel to the brain.Citation6 The brain contains minimal glycogen stores and therefore relies on a consistent supply of glucose for normal function. Counterregulatory hormones are in place to minimise reaching a critical glucose level; these include suppression of insulin secretion at a glucose level of about 4.5 mmol/l, increasing glucagon when glucose level falls below 3.8 mmol/l, and cortisol, growth hormone secretion and upregulation of the sympathetic nervous system function are implemented when level reaches below 3.0 mmol/l.Citation7 Glycaemic thresholds at which these counter-regulatory mechanisms are implemented tend to be lower in diabetic patients and patients with previous hypoglycaemic episodes (see ).Citation6
Figure 3: Glycaemic thresholds.
Chronic hypoglycaemia can result in neurophysiological deterioration in patients with insulinoma with delayed recovery when hypoglycaemia is missed or not rapidly corrected as in this case.Citation8
Peripheral neuropathy as a main presenting complaint in patients with insulinoma is rare, and only described in case reports. It is usually a complication of chronic hypoglycaemia rather than hyperinsulinemia itself.Citation9 The type of neuropathy is usually axonal in nature with both sensory and motor nerves involved. Usually the sensory symptoms recover after removal of the tumour whereas the motor symptoms may persist.Citation9 Seizures are a great mimicker of insulinomas, often misdiagnosed as a primary seizure disorder with electroencephalography findings compatible with epilepsy.
Neurocognitive disorders are common in patients with HIV, occurring in almost half of patients, even in those who are virally suppressed.Citation10 These are related to the virus itself, secondary infections and drug therapies. The presence of this factors might have contributed to the clinical presentation and delay in diagnosis. Neuroendocrine tumours are very rare in HIV-positive patients with only a few case reports published in the literature.
Diagnosis of an insulinoma is based on demonstration of excess endogenous insulin secretion in the setting of hypoglycaemia. Often a 72-hour fast is required to confirm the diagnosis by provoking homeostatic responses in the setting of no caloric intake. The use of biomarkers such as a chromogranin A for diagnostic purposes is not reliable as values maybe be normal in the presence of disease.Citation11
Localisation of the tumour is often a challenge as the majority of insulinomas are small in size and often less than 2 cm. The imaging of choice is still contentious. Often invasive investigations have to be performed in order to localise the tumour. Diagnostic options range from transabdominal ultrasonography to computer tomography, magnetic resonance imaging and nuclear imaging, with MRI having the highest specificity and sensitivity but cost being the limiting factor.Citation12 Transoesophageal endoscopic ultrasonography (TEE) may also be required if the tumour cannot be visualised on imaging.
The treatment of choice in insulinomas is surgical exploration. Medical therapy to manage symptomatic hypoglycaemia is an option for use perioperatively or in a patient who is not a surgical candidate. Medical therapy includes diazoxide (which inhibits insulin secretion) and somatostatin analogues.
This case ended tragically. Unfortunately, because of her severe neurological complications, the patient died prior to any surgical intervention.
Conclusion
Insulinoma is a rare neuroendocrine tumour. Diagnosis is often delayed and can result in lasting neurological complications. Clinicians must always have a higher suspicion of insulinoma in a patient with hypoglycaemia. Investigations need to be expedited to prevent non-reversible neurological consequences of chronic hypoglycaemia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Vortmeyer AO, Huang S, Lubensky I, et al. Non-islet origin of pancreatic islet cell tumors. J Clin Endocrinol Metab. 2004;89(4):1934–8. doi: 10.1210/jc.2003-031575
- Service FJ, McMahon MM, O’Brien PC, et al. Functioning insulinoma--incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66(7):711–9. doi: 10.1016/S0025-6196(12)62083-7
- Placzkowski KA, Vella A, Thompson GB, et al. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987–2007. J Clin Endocrinol Metab. 2009;94(4):1069–73. doi: 10.1210/jc.2008-2031
- Daggett P, Nabarro J. Neurological aspects of insulinomas. Postgrad Med J. 1984;60(707):577–81. doi: 10.1136/pgmj.60.707.577
- Valente LG, Antwi K, Nicolas GP, et al. Clinical presentation of 54 patients with endogenous hyperinsulinaemic hypoglycaemia: a neurological chameleon (observational study). Swiss Med Wkly. 2018;148:w14682.
- Cryer PE. Glucose counterregulation: prevention and correction of hypoglycemia in humans. Am J Physiol. 1993;264(2 Pt 1):E149–55.
- Cryer PE. Hypoglycemia, functional brain failure, and brain death. J Clin Invest. 2007;117(4):868–70. doi: 10.1172/JCI31669
- Pozzessere G, Valle E, D’Alessio C, et al. Effects of spontaneous chronic hypoglycemia on central and peripheral nervous system in insulinoma patients before and after surgery: a neurophysiological follow-up. J Clin Endocrinol Metab. 1997;82(5):1447–51. doi: 10.1210/jcem.82.5.3931
- Tintore M, Montalban J, Cervera C, et al. Peripheral neuropathy in association with insulinoma: clinical features and neuropathology of a new case. J Neurol Neurosurg Psychiatry. 1994;57(8):1009–10. doi: 10.1136/jnnp.57.8.1009
- Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24(9):1243–50.
- Qiao XW, Qiu L, Chen YJ, et al. Chromogranin A is a reliable serum diagnostic biomarker for pancreatic neuroendocrine tumors but not for insulinomas. BMC Endocr Disord. 2014;14:64. doi: 10.1186/1472-6823-14-64
- Noone TC, Hosey J, Firat Z, et al. Imaging and localization of islet-cell tumours of the pancreas on CT and MRI. Best Pract Res Clin Endocrinol Metab. 2005;19(2):195–211. doi: 10.1016/j.beem.2004.11.013