
ABSTRACT
Weaning is known to be the most stressful and critical transition experienced by lambs, it affects the structure of the gastrointestinal tract microbiomes, which significantly influences the lamb’s health and performance. The present study identified the structure of lambs' pre- and post-weaning fecal microbiomes to define the implications of the weaning transition. Illumina MiSeq yielded a total of 5169 sequence reads per fecal sample ranging from 106 to 2028. Before weaning, the taxonomic analysis indicated that the phylum Firmicutes dominated other bacterial phyla at a rate of (≤52.47%), followed by Proteobacteria at a rate of (≤52.00%). On the contrary, the phylum Firmicutes (≤93.01%) tended to increase, while the phylum Proteobacteria tended to decrease (≤26.83 %) after weaning. At the genus level, twenty-eight genera assigned to three phyla were detected in pre-weaned lambs; meanwhile, thirty-three genera assigned to four phyla were identified in post-weaned lambs. These findings suggested that weaning significantly influenced the diversity and the abundance of lambs' fecal microbiota.
1. Introduction
Immediately after birth, microbes from the surrounding environment colonize the gastrointestinal tract of the animal and create a very diverse microbial inhabitant [Citation1]. Ruminant's gastrointestinal tract microbiome has an important function; it converts the stored energy in plant materials into food [Citation2,Citation3]. A well-balanced microbiota, with highly diverse taxonomic content and stability throughout the digestive system, maintains a healthy gut and, consequently, a healthy and productive animal [Citation4].
To face the global challenges of livestock, most studies focused on understanding the rumen microbiomes. However, intestinal microbiomes have a significant role in ruminant early life [Citation5]. Weaning is an important factor that influences the structure of the rumen and intestinal microbiomes. Before weaning, milk microbiota may have an impact on rumen ecology and development. The rumen microbiome would eventually change as a result of weaning and would further be altered due to the stress associated with weaning [Citation6]. Feeding forage to lamb during weaning changed the bacterial population inhabiting the rumen [Citation7]. The gut microbiota of the post-ruminal stage is affected by many factors such as gut motility, pH, host secretions, and redox potential. Most bacteria are lysed before moving into the abomasum due to the low pH and enzymatic activity of the rumen. The microbial abundance and diversity increase in the abomasum, duodenum, and jejunum comparable to the rumen due to the harsh environment [Citation8,Citation9]. In the caecum, colon, and feces, the fermentation conditions are more promising for the microbial growth and diversity in comparison to that in the case of the rumen [Citation10,Citation11].
The study of gastrointestinal microbiota is facing many challenges; the biggest challenge is that most species are not culturable [Citation12]. Currently, whole genome and 16S rRNA gene amplicon sequencing techniques have been used to study the microbiomes of the gastrointestinal tract [Citation13]. The 16S rRNA gene conserved regions are amplified and sequenced using next-generation sequencing. Based on a level of sequence similarity, sequences are assembled into operational taxonomic units (OTUs) [Citation14]. Illumina’s MiSeq sequencing platform recently dominates the next-generation sequencing techniques due to the large data output with accurate contigs in a short time and low costs. Recently next-generation sequencing technology is widely used to study and characterize the changes in the microbiomes of the ruminant's gastrointestinal tract [Citation3,Citation15,Citation16]
Since no study has characterized the implications of the weaning on the gastrointestinal microbiomes in lambs, the present study aimed to identify the shift in the fecal microbiome and the changes which occur as a result of weaning in the lamb's fecal community structure. Furthermore, the establishment of a comprehensive lamb's gut microbiome reference catalogue before and after weaning is addressed.
2. Materials and methods
2.2. Animals understudy and fecal samples collection
The animals of this study were reared and maintained in a private farm following the standard livestock management practices. Lambs were fed on their mother's milk for three months. After weaning, the animals were fed on pellet feed and hay with free access to water and they retained their normal herd behaviour. Fresh pooled fecal samples (5 animals per pool) were collected from pre-weaned (6 days-1 month) and post-weaned (6 months- 12 months) lambs. The samples were placed on ice and sent with a minimal delay to the laboratory, and stored at −80 ° C till further use.
2.3. DNA extraction
Bacterial DNA extraction was carried out using the commercial Quick-DNATM Fecal / Soil Microbe Miniprep Kit (D6010; Zymo Research Corp., Irvine, CA, USA), as directed by the manufacturer. The DNA concentration was measured using a Nanodrop spectrophotometer (Thermo Scientific). For further processing, a final concentration of ∼ 1.8 µg of the extracted DNA was used.
2.4. 16s rRNA amplification and sequencing
Quality control and quantification of the extracted DNA was performed by Genoscreen (Lille, France) with a fluorometric flow. Genoscreen Metabiote approach was used for the preparation of 16S rRNA library, targeting the V3 -V4 region using the universal Forward (TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG) and Reverse (GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATCC) primers. Illumina MiSeq 2 × 250pb run (20000 Reads/samples) was used for 16S rRNA sequencing; the raw sequences were obtained in FASTQ format. 16S rRNA sequences were submitted to the NCBI Sequence Read Archive with the accession number of PRJNA599400 (https://www.ncbi.nlm.nih.gov/sra/PRJNA599400).
2.5. Processing of metagenomic amplicon data
Geneious Prime® 2019.0 (Biomatters Ltd., Auckland, New Zealand) was used for the processing and assembly of raw 16S rRNA reads in the following steps. (1) Trimming and merging paired reads, 16S rRNA amplicon sequences were trimmed, sequences less than 100 bp had been deleted and a region of 250 bp was amplified. The forward and reward reads had merged to create a single consensus sequence. (2) Clustering sequences into OTUs using the de novo assembler, OTUs were defined by sequences clustering by BLAST. Closely-related sequences were clustered into separate contigs using a de novo assembly. (3) Batch blast OTUs and a taxonomy database creation, a curated database was created for the taxonomic classification by blasting the OTUs to the NCBI 16S Microbial database. The BLAST hits were used to create a sequence classifier database. Finally, the extracted Blast-hits were given their source organism name.
2.6. Alpha diversity indices
The alpha diversity of fecal microbial communities was evaluated using three indices (Chao1, Observed Species, and Shannon) which were calculated by QIIME (V1.7.0). Chao1 and Observed Species indices indicate the species richness, while Shannon indicates the abundance, richness, and evenness. Cluster analysis based on the similarity and the dissimilarity of bacterial communities among samples was conducted using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA).
3. Results
3.1. The abundance of OUTs
The fecal microbiota composition of sheep pre- and post-weaning was determined by the 16S rRNA gene (V3- V4 regions) amplicon. Table showed that the total number of raw reads of 16s rRNA (250 bp) was 108,001. A total of 5,169 sequence reads per fecal sample ranging from 106 to 2,028 were retained after quality-filtering with Geneious Prime. All reads that, at 97% identity, passed quality filtering was clustered into OTUs. There were 153 OTUs generated in total, ranging from 12 to 54. The OTUs observed number represents the richness of the sample. The highest number of OTUs was identified in 12- months aged sheep. In general, sheep fecal samples post-weaning had highly abundant OTUs (Figure ).
Figure 1. The number of the observed OTUs clustered at 97% sequence identity.
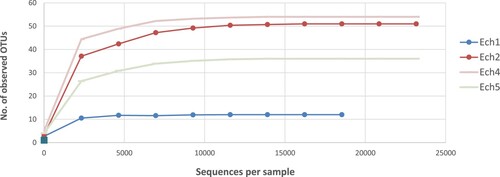
Table 1. The animal characteristics, 16S rRNA amplicon sequence reads and OTUs.
3.2. Fecal microbiomes composition of pre-weaned lambs
Based on the National Center for Biotechnology Information taxonomic database (NCBI) and Geneious Prime analysis, 16S rRNA sequences were assembled into unique OTUs and assigned to different taxonomic levels.
At the phylum level, before weaning, three phyla were detected; Firmicutes Proteobacteria, and Actinobacteria. Ech1 (6-day-old) fecal composition was dominated by Proteobacteria (52.00%), followed by Firmicutes (47.88%), while Ech5 (1-month-old) was dominated by Firmicutes (52.47%), followed by Proteobacteria (47.49). Actinobacteria showed very low abundance rates in Ech1 and Ech5 (0.11%, 0.04%, respectively) Table , Figure .
Figure 2. The relative abundance of the core phyla in the fecal microbiome of pre- and post-weaned lambs.
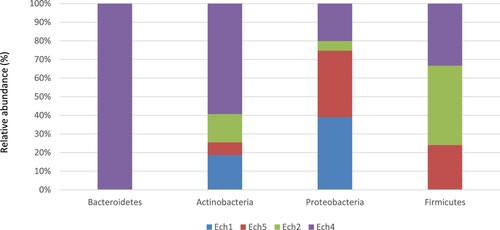
Table 2. Dominant taxa in the fecal microbiota associated with lambs before and after weaning.
At the class level, as shown in Table , five bacterial classes were identified. Clostridia and Bacilli were the most abundant classes before and after weaning, followed by Gammaproteobacteria. On the other hand, class Coriobacteriia, Tissierellia, and Erysipelotrichia were low-abundance classes. Before weaning, the most dominant classes were Clostridia (0.79% in Ech5 and 0.39% in Ech1), Bacilli (0.51% in Ech5 and 0.31%in Ech1), and Gammaproteobacteria (0.11% in Ech5 and 0.48% in Ech1).
At the order level, regardless the age, ten orders were identified, Clostridiales, Enterobacterales, Lactobacillales, Eggerthellales, Bacillales, Tissierellales, Eryspelotrichales, Bifidobacteriales, Selenomonadales, and Coriobacteriales. Clostridiales and Lactobacillales were defined as the core orders. Before weaning, the most dominant orders were Clostridiales (0.90% in Ech5 and 0.39% in Ech1), Lactobacillales (0.11% in Ech5 and 0.24% in Ech1), and Enterobacteriales (0.06% in Ech5 and 0.39% in Ech1). However, Coriobacteriales and Tissierellales were absent in Ech5 and Ech1.
At the family level, nineteen families were identified, including Clostridiaceae, Enterococcaceae, Lachnospiraceae, Erwiniaceae, Eggerthellaceae, Bacillaceae, Peptostreptococcaceae, Streptococcaceae, Enterobacteriaceae, Aerococcaceae, Ruminococcaceae, Carnobacteriaceae, Bifidobacteriaceae, Tissierellaceae, Erysipelotrichaceae, Oscillospiraceae, Rikenellaceae, Bacteroidaceae, and Atopobiaceae. The families Clostridiaceae, Enterococcaceae, and Lachnospiraceae were defined as the core families before and after weaning. Before weaning, the most dominant families were Clostridiaceae (0.17% in Ech1 and 0.23% in female), Enterococcaceae (0.11% in Ech1 and 0.24% in Ech5), Lachnospiraceae (0.17% in Ech1 and 0.15% in Ech5) and Enterobacteriaceae (0.17% in Ech1 and 0.24%in Ech5). It is noteworthy that Aerococcaceae, Ruminococcaceae, Carnobacteriaceae, Bifidobacteriaceae, Tissierellaceae, Oscillospiraceae, Rikenellaceae, Bacteroidaceae and Atopobiaceae were absent in both Ech1 and Ech5.
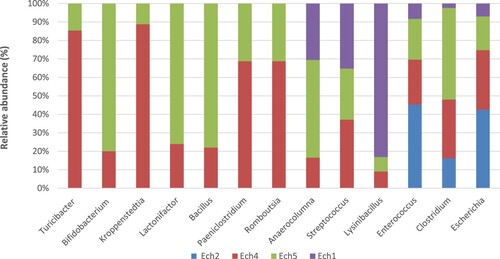
At the genus level, 48 genera were identified according to Table and Figure . Of these, 13 genera were found before and after weaning including Escherichia (8.49% to 51.39%), Clostridium (0.94% to 19.38%), Enterococcus (1.89% to 10.33%), Lysinibacillus (8.33% to 76.42%), Streptococcus (2.22% to 2.98%), Anaerocolumna (0.51% to 1.63%), Romboutsia (0.05% to 0.11%), Paeniclostridium (0.05% to 0.11%), Bacillus (0.11% to 0.39%), Lactonifactor (0.11% to 0.35%), Kroppenstedtia (0.10% to 0.79%), Bifidobacterium (0.11% to 0.44%) and Turicibacter (0.25% to 1.46%). Before weaning, 15 genera were found including Vagococcus, Shigella, Eggerthella, Anaerobacillus, Anaerostipes, Erwinia, Desnuesiella, Paraclostridium, Pseudoflavonifractor, Lachnotalea, Hydrogenoanaerobacterium, Urmitella, Fonticella, Alkalibacter, and Paenibacillus (Figure ). It is worthy mention that the top core genera Escherichia and Clostridium are mainly responsible for the neonatal diarrhea.
Figure 3. The relative abundance of the core genera in the fecal microbiome of pre- and post-weaned lambs.
3.3. Fecal microbiomes composition of post-weaned lambs
At the phylum level, four phyla were detected; the phylum Firmicutes dominated the bacterial communities, followed by Proteobacteria, Actinobacteria, and Bacteroidetes. Ech2 fecal composition (6-month-old) was dominated by Firmicutes (93.01%) and Proteobacteria (6.88%). In Ech4 (12-month-old), Firmicutes (72.76%) and Proteobacteria (26.83%) were the most abundant phyla. Actinobacteria showed very low abundance rates in both Ech2 and Ech4 (0.09%, 0.35%, respectively), while the phylum Bacteroidetes was detected only in Ech4 at a rate of (0.06%) (Table ), (Figure ).
At the class level, Table showed the prevalence of six bacterial classes; Clostridia (4.71% in Ech2 and 0.88% in Ech4), Bacilli (3.77% in Ech2 and 0.54% in Ech4), and Gammaproteobacteria (0.94% in Ech2 and 0.10%in Ech4) were the dominant classes. The class Tissierellia was only found in Ech4 (12-month-old).
At the order level, after weaning, the most dominant orders were Clostridiales (4.71% in Ech2 and 0.88% in Ech4), Lactobacillales (2.83% in Ech2 and 0.05% in Ech4), and Bacillales (0.94% in Ech2 and 0.25% in Ech4). Whereas, Eggerthellales and Selenomonadales were absent in both Ech2 and Ech4. Clostridiales and Lactobacillales were significantly more abundant in Ech2.
At the family level, the families Clostridiaceae, Enterococcaceae, and Lachnospiraceae were defined as the core families before weaning. Similarly, after weaning, the most abundant families were Clostridiaceae (3.77% in Ech2 and 0.15% in Ech4), Enterococcaceae (1.88% in Ech2 and 0.05% in Ech4), Lachnospiraceae (0.94% in Ech2 and 0.25% in Ech4), Bacillaceae (0.94% in Ech2 and 0.19% in Ech4), Peptostreptococcaceae (0.94% in Ech2 and 0.25% in Ech4), and Streptococcaceae (0.94% in Ech2 and 0.10% in Ech4). Interestingly, both Erwiniaceae and Eggerthellaceae were not found in both Ech2 and Ech4.
At the genus level, Table and Figure showed that in addition to the 13 core genera, 20 genera were found after weaning including Terrisporobacter, Sporobacter, Anaerotignum, Mageeibacillus, Muricomes, Tissierella, Alistipes, Faecalicatena, Intestinimonas, Atopobium, Gracilibacter, Bacteroides, Eubacterium, Lachnoclostridium, Butyricicoccus, Mogibacterium, Abiotrophia, Papillibacter, Coprococcus, and Oscillibacter.
3.4. Microbial community richness and diversity
Chao1, Species Observed, and Shannon indices were used to analyze the species diversity of the fecal samples. The indices revealed that the highest level of species complexity was found in weaned lambs, where a total of sixty-five species was revealed (Figure ), while the lowest level of complexity was found in pre-weaned lambs, where only fifty-five species were found (Figure ). The result of the group α-diversity analysis identified 12-months aged lambs as more complex concerning their species diversity (Table ), (Figure ).
Figure 4. Phylogenetic tree built with the Geneious Tree Builder using the unweighted pair group method with arithmetic mean (UPGMA) showed the species-level composition of the fecal microbiome in post-weaned lambs.

Figure 5. Phylogenetic tree built with the Geneious Tree Builder using the unweighted pair group method with arithmetic mean (UPGMA) showed the species-level composition of the fecal microbiome in pre-weaned lambs.

Figure 6. Richness (Chao-1), and evenness (Shannon evenness) indexes observed in pre-and post- weaned lambs.
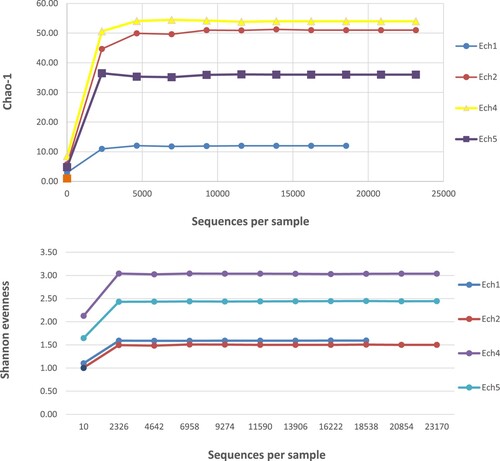
Table 3. Alpha diversity metrics.
4. Discussion
The development of a lamb’s gastrointestinal microbiomes begins at birth, while the most significant change occurs at weaning transition [Citation5]. In this study, we used next-generation sequencing technology to characterize the shift in the compositions of lamb's fecal microbiota before and after weaning. Our results suggested that lambs’ fecal microbiota had a greater diversity and richness after weaning. This was in line with an earlier study on calves that showed that the introduction of solid feed post-weaning was expected to promote rumen bacterial diversity [Citation17]. Our study was also in line with a study on the composition of the rumen microbiome of goats at different ages; which concluded that the rumen microbiome of lambs is different from that of adult goats [Citation3].
Young ruminants fed on milk which is digested in the abomasum and acts as monogastric until their digestive system is fully developed. The development of the reticulo-rumen and its microbial inhabitants is required for the transition to a ruminant, with a slight loss in their growth, and better utilization of solid diets [Citation18]. The development of the rumen has three phases: from 0 to 3 weeks old is the non-rumination phase, from 3 to 8 weeks old the transitional phase, and from 8 weeks on is the rumination phase [Citation19]. It has been shown that rumen development is incomplete at 12 weeks of age in young ruminants that have been fed on milk [Citation20]. At birth, the gastrointestinal tract of mammals’ animals is germ-free. The colonization started after the neonate is born and ingested microbes form the surrounding environment. The predominant bacteria after birth belong to the phylum Proteobacteria, especially E. coli. However, milk consumption changes the gut microbiota to lactic acid bacteria [Citation21]. The development of the rumen microbiota is slow in the young ruminant. The bacterial community found in the non-mature rumen showed that the major types of rumen bacteria are at the age of 14 days including cellulolytic and proteolytic species [Citation22]. One day after birth, some of the ruminal microbiota which is critical for rumen function might be detected. The establishment of the ruminal bacterial community in ruminants has been detected from birth to weaning [Citation23]. After birth, the establishment of bacteria is exponentially fast and sequential. The phylum Bacteroidetes is replacing the phylum Proteobacteria as the main Phyla. The developed rumen was comprised of various bacteria between 3 and 12 days. Before the ingestion of solid food starts, the bacteria responsible for food fermentation are present. The diet influence appeared stronger and was related to an alteration in the bacterial inhabitants between days 9 and 15. From 15 days on, changes in the relative richness of some genera occurred [Citation24].
After weaning, fecal microbiota had a greater richness and evenness, this may be due to the increase of the substrates after the higher intake of solid feed. The greater availability of substrates and the stability of the gut pH encourage the diversity of the bacterial community [Citation25]. In agreement with earlier studies on cattle [Citation5,Citation16], our results revealed that Firmicutes (>93.01%) and Proteobacteria (>52.00%) were the most abundant phyla. The abundance of the Firmicutes phylum was significantly higher compared to other phyla, especially the order Clostridiales. After weaning, the phylum Firmicutes tended to increase, but the phylum Proteobacteria tended to decrease. A next-generation sequencing study in 14 days ruminant animals showed that the genera under the phyla Bacteroidetes, Firmicutes, and Proteobacteria rated 98%, targeting the (V3-V5) of 16S rRNA gene [Citation22]. In 12 months of age, there is a decrease in the phyla Proteobacteria and an increase in the phyla Bacteroidetes. Moreover, at the age of 14, 42 days, and 12 months, the Proteobacteria in the rumen microbiota accounted for > 20%. Regardless of the age at weaning, Meale reported a decline in the abundance of the phylum Bacteroidetes before weaning and an increase in the phylum Firmicutes after weaning, in conjunction with the abundance of the phylum Proteobacteria [Citation5]. These results were consistent with previous studies that confirmed the dominance of phyla Bacteroidetes, Firmicutes, and Proteobacteria after weaning [Citation26,Citation27]. Conversely, Li et al. studied the effect of early weaning on the intestinal microbiota in lambs and found that the abundance of Proteobacteria significantly increased with early weaning; meanwhile, the abundance of Firmicutes significantly decreased. It seems that early weaning had a substantial effect on the intestinal microbiota in lambs [Citation22]. Han et al. observed in the 100-day-old lambs, the phylum Proteobacteria was significantly higher than in the 80 and 90-day-old lambs [Citation3]. Interestingly, although the differences in bacterial diversity among different ages were not significant, the abundance of fecal microbiota changed with age. These findings were in an agreement with earlier observations in ruminants [Citation28]. Similarly, a study explored the bovine rumen bacterial community from birth to adulthood [Citation16], Firmicutes, Bacteroidetes, and Proteobacteria were the core phyla found in all age groups with a varied abundance of genera. We suspected that the fecal bacterial composition may differ with the maturation of the lambs. It should be noted that the age difference among lambs in this study is about 4–5 months. Possibly the bigger age gap will reflect more differences in bacterial diversity, which requires further study.
At the genus level, the core genera at all ages were Escherichia (>51.39%), Clostridium (>19.38%), Enterococcus (>10.33%), Lysinibacillus (>76.42%), Streptococcus (>2.98%), Anaerocolumna (>1.63%), Romboutsia (>0.11%), Paeniclostridium (> 0.11%), Bacillus (0.11% to 0.39%), Lactonifactor (>0.35%), Kroppenstedtia (> 0.79%), Bifidobacterium (>0.44%) and Turicibacter (> 1.46%). The most abundant genera Escherichia, Clostridium, Enterococcus decreased with age. Interestingly, twenty-eight genera were detected during the primary stages of colonization, while thirty-three genera were detected in mature animals. An earlier study suggested that the microbiota was more diverse at 84 days than at 42 days of age [Citation22]. Theoretically, “functional redundancy” provides by a high level of diversity which allows an ecosystem to be stable and resilient to environmental stressors [Citation29]. Generally, the highly diverse gut microbiota is considered beneficial for host health and considered also as a sign of mature gut microbiota [Citation30,Citation31]. On the other hand, some studies have found that the development and diversification of the microbiota may have a negative impact on the immune function [Citation32]. In this study, Proteobacteria significantly increased in the abundance before weaning. The phylum Proteobacteria have a wide variety of pathogens, such as Escherichia, Salmonella [Citation33], we also found that before weaning the genus Escherichia coli significantly increased in the abundance.
It has been thought that the genus Clostridium has to be closely related to diarrhea in ruminants. Clostridium has toxins that affect the body through different pathways [Citation34]. The high abundance of the genus Clostridium among lambs in the current study may explain the softness of the feces consistency and why lambs frequently develop diarrhea. It has been found in some studies that the abundance of Turicibacter increases during enteritis, however, Turicibacter pathogenicity has not been clarified [Citation35]. Furthermore, it has been reported that, in intestinal diseases, an increase of urease enzymes and bile salt hydrolase provides an environment for the survival of Romboutsia [Citation36]. This conveys a message that a reduction in beneficial bacteria leads to a pathogen invasion. Though, the genera Paeniclostridium, Turicibacter, and Romboutsia were in a minor abundance in this study.
Inevitably, the health state is related to ages. Lambs have an extremely high rate of diarrhea, and this condition improves with age. Moreover, the intestinal microflora has been linked to diarrhea [Citation37]. However, the microbiome is an extremely fragile ecosystem. We suspect that the gut microbiome may not be the only reason why lambs are more likely to have diarrhea than adult sheep. Accordingly, other factors such as low pH in the gastric, intestinal developmental immaturity, changes in feed components, and some the external environment may make the lambs more susceptible to the invasion of the pathogen [Citation38]. The foreign pathogen invasion causes the lamb to develop diarrhea, consequently, dynamic changes in the intestinal microbiota caused by the pathogen. After weaning, bacteria that were established during breast-milk feeding are gradually replaced by the bacteria associated with the digestion of food typically, weaning combines many physical and psychological stressors that can increase susceptibility to disease and change the immune state. Besides, the study has shown that weaning age may influence the inflammatory response of the lower gut [Citation5]. No strong conclusions could be drawn in that regard from that research because this was not specifically studied.
In agreement with a previous study on calves [Citation5], the Shannon evenness index was different in the fecal samples of the pre-and post-weaned lambs, as were measures of OTUs richness indices, Chao1 and the observed OTU (Table ) (Figure ). The fecal microbiota of pre-weaned lambs was less diverse and evenness, whereas fecal microbiota of post-weaned lambs had a greater richness and evenness.
5. Conclusion
In conclusion, weaning has a significant influence on the richness and the diversity of the lamb's fecal microbiomes. A clear understanding of the composition and diversity of the gastrointestinal tract microbiome is required to improve the ruminant's growth and health. Characterization of the gastrointestinal tract bacterial composition at the species level is important to detect the pathogens in small ruminants.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Fernández HM, Elisabeth S. A complete estimate of the phylogenetic relationships in Ruminantia: a dated species-level supertree of the extant ruminants. Biol Rev. 1999;80(2):269–302. doi: 10.1017/S1464793104006670
- Reece WO. Functional anatomy and physiology of domestic animals. Baltimore: Lippincott Williams & Wilkins; 2005; p. 357–358.
- Han X, Yang Y, Yan H, et al. Rumen bacterial diversity of 80 to 110-day- old goats using 16S rRNA sequencing. PLOS ONE. 2015;10(2):e0117811. doi: 10.1371/journal.pone.0117811
- Tilg H, Kaser A. Gut microbiome obesity and metabolic dysfunction. J Clin Invest. 2011;121:2126–2132. doi: 10.1172/JCI58109
- Meale SJ, Li SC, Azevedo P, et al. Weaning age influences the severity of gastrointestinal microbiome shifts in dairy calves. Sci Rep. 2017;7:198. doi: 10.1038/s41598-017-00223-7
- Yáñez-Ruiz DR, Macías B, Pinloche E, et al. The persistence of bacterial and methanogenic archaeal communities residing in the rumen of young lambs. FEMS Microbiol Ecol. 2010;72:272–278. doi: 10.1111/j.1574-6941.2010.00852.x
- Jeyanathan J, Martin C, Morgavi DP. The use of direct-fed microbials for mitigation of ruminant methane emissions: a review. Animal. 2014;8:250–261. doi: 10.1017/S1751731113002085
- He J, Yi L, Hai L, et al. Characterizing the bacterial microbiota in different gastrointestinal tract segments of the Bactrian camel. Sci Rep. 2018;8:654. doi: 10.1038/s41598-017-18298-7
- Yeoman CJ, Ishaq SL, Bichi E, et al. Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci Rep. 2018;8:3197. doi: 10.1038/s41598-018-21440-8
- De Oliveira MNV, Jewell KA, Freitas FS, et al. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet Microbiol. 2013;164:307–314. doi: 10.1016/j.vetmic.2013.02.013
- Popova M, Mcgovern E, Mccabe MS, et al. The structural and functional capacity of ruminal and cecal microbiota in growing cattle was unaffected by dietary supplementation of linseed oil and nitrate. Front Microbiol. 2017;8:937. doi: 10.3389/fmicb.2017.00937
- Siezen RJ, Kleerebezem M. The human gut microbiome: are we our enterotypes? Microb Biotechnol. 2011;4:55053.
- Wells JE, Kim M, Bono JL, et al. Escherichia coli O157:H7, diet, and fecal microbiome in beef cattle. J Anim Sci. 2014;92(4):1345–1355. doi: 10.2527/jas.2013-7282
- Shafquatm A, Joice R, Simmons SL, et al. Functional and phylogenitic assembly of microbial communities in the human microbiome. Trends Microbiol. 2014;22(5):261-6.
- Li RW, Conner EE, Li C, et al. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbiol. 2012;14:129–139. doi: 10.1111/j.1462-2920.2011.02543.x
- Jami E, Israel A, Kotser A, et al. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013;7:1069–1079. doi: 10.1038/ismej.2013.2
- Krause DO, Khafipour E. The fecal environment, the gut. In: MJ Sadowski, RL Whitman, editor. The FecalBacteria. Washington, DC: ASMPress; 2010. p. 1–21.
- Heinrichs J. Rumen development in the dairy calf. Adv Dairy Technol. 2005;17:179–187.
- Lane M, Baldwin R, Jesse B. Developmental changes in ketogenic enzyme gene expression during sheep rumen development. J Anim Sci. 2002;80:1538–1544. doi: 10.2527/2002.8061538x
- McCann JC, Wickersham TA, Loor J. High-throughput methods redefine the rumen microbiome and its relationship with nutrition and metabolism. Bioinform Biol Insights. 2014;8:109–125. doi: 10.4137/BBI.S15389
- Varel VH, Wells JE. Lower digestive tract microbiology. In: WG Pond, AW Bell, editor. The encyclopedia of animal science. New York, NY: Marcel Dekker, Inc; 2005. p. 585.
- Li C, Wang W, Liu T, et al. Effect of early weaning on the intestinal microbiota and expression of genes related to barrier function in lambs. Front Microbiol. 2018;9:1431. doi: 10.3389/fmicb.2018.01431
- Rey M, Enjalbert F, Combes S, et al. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J Appl Microbiol. 2014;116:245–257. doi: 10.1111/jam.12405
- Yáñez-Ruiz DR, Abecia L, Newbold CJ. Manipulating rumen microbiome and fermentation through interventions during early life: a review. Front Microbiol. 2015;6:1133. doi: 10.3389/fmicb.2015.01133
- Khafipour E, Li S, Plaizier JC, et al. Microbiome analysis of the rumen, cecum, and feces of dairy cows with subacute ruminal acidosis. J Anim Sci. 2011;89:489. doi: 10.2527/jas.2009-2458
- Baldwin RL, Vi McLeod KR, Klotz JL, et al. Rumen development, intestinal growth and hepatic metabolism in the pre-and postweaning ruminant. J Dairy Sci. 2004;87:55–65. doi: 10.3168/jds.S0022-0302(04)70061-2
- Dehority BA. Rumen microbiology. Nottingham: Nottingham Univ. Press; 2003; P. 307.
- Wang J, Fan H, Han Y, et al. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Aus J Anim Sci. 2017;30:100–110. doi: 10.5713/ajas.16.0166
- Konopka A. What is microbial community ecology? ISME J. 2009;3:1223–1230. doi: 10.1038/ismej.2009.88
- Turnbaugh PJ, Ley RE, Hamady M, et al. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244
- Chatelier L, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506
- Nylund L, Satokari R, Nikkila J, et al. Microarray analysis reveals marked intestinalmicrobiota aberrancy in infants having eczema compared to healthy childrenin at-risk for atopic disease. BMC Microbiol. 2013;13:12. doi: 10.1186/1471-2180-13-12
- Chen L, Xu Y, Chen X, et al. The maturing development of gut microbiota in commercial piglets during the weaning transition. Front Microbiol. 2017;8:1688. doi: 10.3389/fmicb.2017.01688
- Lewis CJ, Naylor RD. Sudden death in sheep associated with clostridium sordellii. Vet Rec. 1998;142:417–421. doi: 10.1136/vr.142.16.417
- Bretin A, Lucas C, Larabi A, et al. AIEC infection triggers modification of gut microbiota composition in genetically predisposed mice, contributing to intestinal inflammation. Sci Rep. 2018;8:12301. doi: 10.1038/s41598-018-30055-y
- Gerritsen J, Hornung B, Renckens B, et al. Genomic and functional analysis of Romboutsia ilealis CRIBT reveals adaptation to the small intestine. PeerJ. 2017;5:3698. doi: 10.7717/peerj.3698
- Türkyılmaz S, Eskiizmirliler S, Tunaligil S, et al. Identification, characterization and molecular epidemiology of Escherichia coli isolated from lamb and goat kids with diarrhoea. Acta Vet Brno. 2013;82:357–362. doi: 10.2754/avb201382040357
- Causapé AC, Quıìlez J, Sánchez-Acedo C, et al. Prevalence and analysis of potential risk factors for Cryptosporidium parvum infection in lambs in Zaragoza (Northeastern Spain). Vet Parasitol. 2002;104:287–298. doi: 10.1016/S0304-4017(01)00639-2