
Abstract
Carbonyl reductase 1 (CBR1), a short-chain dehydrogenase/reductase, is important in the phase I reduction of drugs. Glutathione (GSH) facilitates the access of low-affinity substrates to the CBR1 active site. This study investigates the path of substrates to the CBR1 active site when GSH occludes the entrance to NADP and binds its catalytic residues. A set of CBR1 structures bound with GSH were subjected to comprehensive docking and molecular dynamics (MD) simulations. The glycine moiety of GSH dangled within the active site, converting the active site into ON/OFF status, while the glutamate residue slips into a hydrophobic cavity, allowing more room for driving the substrate toward the active residues. GSH spontaneously moves within the apoCBR1 binding site and acts as an internal control barrier that selects substrates entering the catalytic site. The VAL230-TYR251 was most flexible in ApoCBR1 structure and showed a lower degree of flexibility upon the binding of inhibitors.
1. Introduction
Drug metabolism passes through two main phases, phase 1 and phase 2. In phase 1, polar groups are added to the drugs to render it more water-soluble and comprise oxidation, reduction or hydrolysis. Phase 2 comprises conjugation with charged molecules as GSH or glucuronic acid [Citation1]. CBR1 is a short-chain reductase/dehydrogenase that reduces a wide variety of compounds using NADH as a coenzyme [Citation2,Citation3]. CBR1 is an important member of phase I drug metabolism by reduction of compounds and drugs containing carbonyl group [Citation3]. In humans, there are two isoforms of CBRs, CBR1 & CBR3, with approximately 85% sequence similarity [Citation4]. Although the exact pharmacological role of CBR1 has not yet been fully elucidated, it acts on diverse substrates, i.e. 1,2-naphthoquinone [Citation5], 2,3-butanedione [Citation6], 1,4-naphthoquinone [Citation5], 3-glutathionyl-4-hydroxynonanal [Citation7], doxorubicin [Citation8], duroquinone [Citation9], indole 3-acetaldehyde and ubiquinone-1 [Citation9], oracin [Citation5]. In addition to its classic function in maintaining body homeostasis, CBR1 reductive activity resulted in the induction of cardiotoxicity by anthracycline anticancer drugs through the formation of cardiotoxic C-13 hydroxyl derivatives Wermuth. Interestingly, 13-deoxy-anthracyclines, which lack the C-13 hydroxyl group to prevent their metabolism by CBR1, are more potent anticancer agents with lower toxicity than their parent compounds [Citation10]. Inhibition of CBR1 is important in handling chemo-resistance and cardiotoxicity of the anticancer agents doxorubicin and daunorubicin [Citation11,Citation12].
A glutathione (GSH) binding site was discovered in CBR1. It is believed that poorly recognized substrates can conjugate with the thiol group of the cysteine moiety of GSH, allowing them to be better recognized by the CBR1 active site [Citation13]. In this context, GSH has been shown to bind to the active site of CBR1, including two of its catalytic triad residues [Citation14]. This binding may hinder the entrance of substrates to the active site or turns it off.
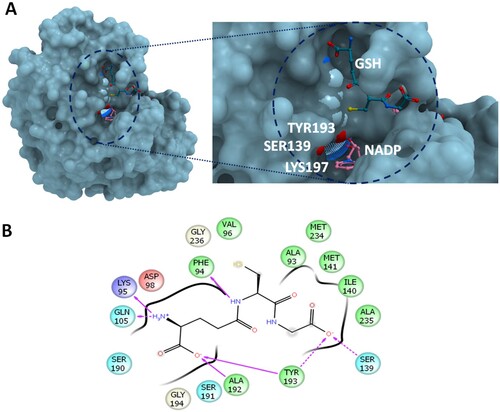
The catalytic triad involved in CBR1 activity consists of Ser139, Tyr193 and Lys197. Ser139 stabilizes the CBR1 substrates by interacting with their carbonyl group, TYR193 is the catalytic proton donor, while LYS197 lowers the pKa value of TYR-OH and promotes donor transfer by forming hydrogen bonds with the nicotinamide ribose moiety [Citation15]. Glutathionylated substrates were found to bind to a specific GSH binding site (Figure (A)) close to the active site of CBR1 [Citation14,Citation16]. GSH adducts have been shown to be essential for proper metabolism of some substrates, such as PGA1, but did not affect the reduction rate of other substrates, i.e. menadione [Citation7]. GSH protects the binding sites from entering the nonspecific substrates and allows only substrates with sufficiently high affinity and capable of displacing GSH to its “ON” conformation [Citation14]. Besides, GSH facilitates the entrance of poor-binding substrates to the active site for further catalysis by CBR1 [Citation17]. The recognition of hydrophobic substrates in the binding site of CBR1 is facilitated by residues TRP229, ALA235 and MET241 [Citation18].
Figure 1. The molecular interactions at the active site of human CBR1. (A) GSH in open conformation bound with CBR1. The reactive groups of S139 and Y193 were shown in red (B) The ligand interactions of GSH before of MD simulation. The interaction coordinates were retrieved from the crystal structure 4Z3D. Hydrophobic residues are displayed in green; positive charged, in blue; and negative charged, in red. The direction of arrows indicates the hydrogen bond donor-receiver. Dotted arrows indicates interaction with the backbone of protein residues, while solid-line arrows indicates interaction with the side chains atoms.
The exact mechanism of GSH acting as a molecular switch for the active site of CBR1 is not well understood. Thus, utilizing molecular dynamics (MD) simulation, a meta-analysis of co-crystallized ligands was performed to assess the molecular switch features in detail. MD is a standard method for understanding the structural changes in response to different ligands. The major objectives of this study are to explore the substructural changes during metabolic switch formation. The position and conformation of GSH will be monitored during MD simulation. Besides, GSH interactions with the catalytic residues will be mapped. Additionally, the influence of GSH on MD parameters of low-affinity ligands such as 2,3-butanedione was evaluated [Citation6]. The anticipated results will help design specific CBR1 inhibitors, efficiently characterize the mechanisms associated with drug metabolism and evaluate CBR1 responses to low-molecular-weight (mw) substrates with poor affinity in the presence and absence of GSH.
2. Materials and methods
2.1. Protein preparation
The structures of CBR1 were retrieved from the Protein Data Bank (PDB, Table ). These structures include CBR1 in complex with hydroxyl-PP (PDB ID 1WMA), (PDB ID 2PFG), GSH in protective configuration (PDB ID 4Z3D), NADP (PDB ID 3BHI), GSH (3BHJ) and S-hydroxymethylglutathione (3BHM).
Table 1. The molecular targets used in this computational study.
The PDB IDs displayed in Table were retrieved from the PDB. Initially, the structure was corrected for errors in side chains, missing residues or missing atoms by using the protein preparation wizard implemented in Maestro version 12 (Schrodinger LLC, NY, USA) [Citation19]. Only the protein structures and substrates were included in the work space, and then charges and nonpolar hydrogens were added and the protein was optimized at physiological pH. Energy minimization was carried out by OPLS2005 force field. The missing charges, atoms or residues were modelled in Schrodinger package.
2.2. Molecular dynamics simulation
MD simulation was employed to assess the substructural changes associated with the presence of GSH and various substrates linked with GSH binding. After initial preparation, MD simulation was executed in YASARA software (version 14.12.2) by using the AMBER14 force field implemented in the YASARA structure [Citation20–22]. The particle-mesh Ewald algorithm was used for handling long-range electrostatic force. The simulation was carried out under the NPT ensemble of isothermal (298 K) and isobaric conditions for 100 ns. A simulation cell was constructed and loaded with water (0.997 g/mL density). To mimic physiological conditions, Na or Cl was added at a concentration of 0.9%. The distance between the protein and the cell wall was set to 10 Å. The time step was set to 2 fs, and simulation snapshots were collected every 100 ps. Steepest descent minimization was used to initialize the structure preparation for long MD experiments. MD was run by a predefined macro implemented in YASARA software (MD_runfast.mcr). The MD_analyse and MD_analyseres macros were used to analyse energy and residual component changes during the MD run. The MD simulation was used to analyse the RMSD changes, energetic changes as well as the per residue changes of CBR1 bound with GSH and other substrates. Comparisons were made based on the generated average structure from three experiments.
2.3. Molecular docking
To evaluate the effect of GSH on low-mw low-affinity substrates, a docking run followed by MD simulation was designed. For this purpose, a low-affinity ligand was selected from the available literature. 2,3-Butanedione is a low-affinity CBR1 ligand with Km = 1.8 mM and kcat = 6.2 s−1 [Citation6]. The 2D conformation of 2,3-butanedione was retrieved from the PubChem database, followed by 3D optimization by Chem3D CambridgeSoft software.
Docking was carried out with Molegro Virtual Docker 5.5 software by using 4Z3D structure (Table ). MolDock was used as a scoring function. Docking was followed by assigning a template file from GSH bound with active site residues. Docking was allowed only within 5 Å of the selected template position. The generated docking template was used in a docking run to deliver 10 poses. The validity of docking was confirmed by the redocking of the co-crystallized ligand. The generated docked CBR1-2,3-butanedione complex was saved as a PDB file and used for the MD experiment. MD parameters and specifications were as described above.
2.4. Other molecular modelling methods
Handling, visualization and generation of figures were executed by employing multiple software packages, including ICM molsoft, Maestro, Microsoft Excel and GraphPad Prism software.
3. Results
3.1. GSH interactions
While GSH forms a network of hydrogen bonding interactions with CBR1, the main contributor to this network is the GSH-glutamate moiety and, to a lesser extent, the GSH-glycine moiety. Figure shows the ligand interactions of GSH with the side chains of CBR1. The glutamate moiety forms hydrogen bonds with Ala192, Tyr193, Lys95 and Gln105 (Figure ). The hydroxyl group of the glycine moiety of GSH forms hydrogen bonds with CBR1 TYR193 and SER139 of the catalytic triad. Before simulation, the glycine residue of GSH is projecting toward the catalytic portion or the nicotinamide ring of NADPH (Figure (A)).
Figure 2. Simulation trajectories at different times during 100 ns of MD simulation. After the start of simulation, the simulation trajectories were displayed in YASARA software under the macro MD-play followed by export of screenshots. GSH and NADP are displayed as green sticks. CBR1 is displayed as a surface representation. Blue indicates a positively charged surface, red indicates negatively charged residues, and white indicates neutral residues.
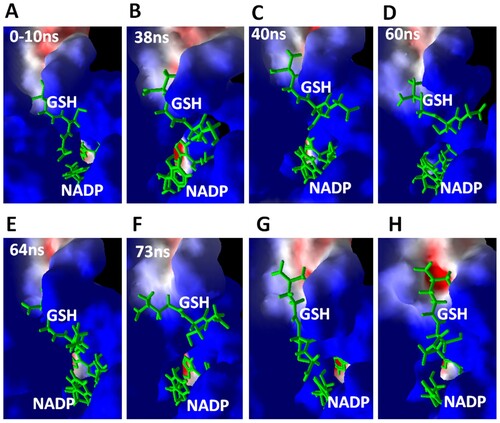
3.2. Dynamic changes in GSH during MD simulation
Figure shows simulation trajectories at different times during the MD simulation. These simulation trajectories illustrate the changes in GSH conformation during the MD simulation and supported by Supplementary Video 1. The trajectories obtained after the MD simulation were displayed in YASARA software by running the macro MD-play followed by the export of simulation trajectories at the displayed times. The typical conformation and interaction of GSH within the crystal structure (4Z3D) were conserved during the first 10 ns of simulation (Figure (A)), where the glycine moiety of GSH forms “OFF” conformation by closing the access to the active site. The carboxyl group of the glycine moiety is projecting toward the aperture, leading to NADPH at the active site and forming hydrogen bonds with two residues of the catalytic triad, SER139 and TYR193 (Figure (A,B)). After 20 ns, the glycine moiety shows marked deviation from its binding site, exposing the active site residues and opening the aperture to NADPH (Figure (B–F)). At 82 ns of simulation, GSH adopts an extended conformation, and the glycine moiety turns off the active site again (Figure (G,H)). Therefore, GSH spontaneously turns from off to on position during simulation.
The conformations of GSH detected during MD simulation indicate interesting changes in the glycine and glutamate moieties of GSH. The glycine moiety turns the active site on and off; by moving to the right, away from the catalytic residues SER139 and TYR193, the active site is open to new ligands. On the other hand, moving to the left resulted in binding with the catalytic residues SER139 and TYR193 and the active site remaining closed for new ligands. Additionally, the glutamate residue slips into the cavity at the upper part of the active site formed by the hydrophobic residues VAL96-THR109. This movement might enable beneficial adaptation to ligands of various sizes.
3.3. Structural stability of CBR1 complexes
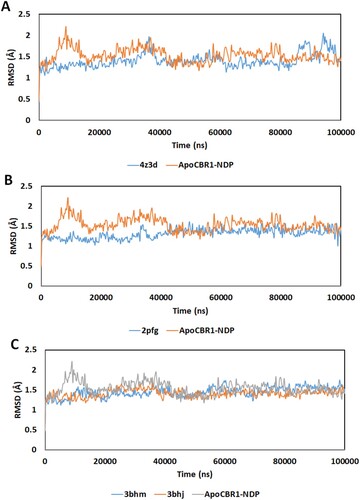
The RMSD of the α-carbon atom in the CBR1 structures during 100 ns of MD simulation is shown in Figure . RMSD was calculated after the MD run by computing the average distance of the α-carbon in the protein backbone by superimposition of CBR1 at the start and end of simulation. CBR1 bound to NADP without any other substrate (ApoCBR1-NDP) reached stability at approximately 20 ns with an average RMSD of 1.5 Å (Figure (A)). The binding of GSH decreased the average RMSD value (4Z3D). However, several drifts in RMSD are observed, which might be attributed to the changes associated with the movement of the glycine moiety of GSH during the MD simulation. The structures containing modified GSH, including 2PFG, 3BHM and 3BHJ (Figure (B,C)), showed almost the same profiles of continued stability starting from the first few ns of simulation. The drifts observed with 4Z3D (nonmodified GSH) were not observed with the other structures. The structures 3BHM and 3BHJ contain the substrate mimic OH-PP at the binding site. This provided constraints on the GSH movement observed with GSH alone.
Figure 3. RMSD of the α-carbon atom in the CBR1 structures during 100 ns of MD simulation. (A) Apo-NADP: CBR1 bound with NADP. 4Z3D: CBR1 bound with GSH in closed form and NADP. (B) 2PFG: CBR1 bound with BiGF2 and NADP. (C) 3BHJ: CBR1 bound with GSH, OH-PP and NADP. 3BHM: CBR1 bound with S-hydroxymethylglutathione and NADP.
3.4. Residual changes and fluctuations
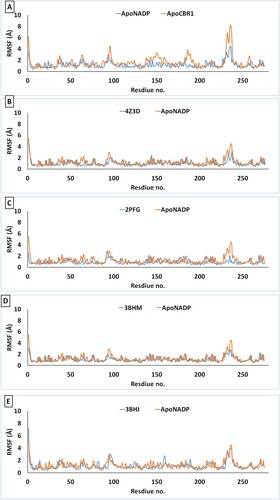
The changes in RMSF for every residue of the protein are shown in Figure . These values were calculated by running the YASARA macro MD-analysres. Per-residue changes in RMSF reveal two positions of high flexibility. The first is the VAL230-TYR251 loop, which contains residues important for binding of NADP as well as substrate binding. The binding of NADP stabilizes this loop (Figure (A)). Compared to ApoCBR1, the VAL230-TYR251 loop exhibits decreased RMSF when it binds with NADP. The second flexible loop, ILE92-PHE102, showed almost conserved changes in all structures.
Figure 4. Per-residue RMSF of CBR1 structures during 100 ns of MD simulation. (A) Apo-NADP: CBR1 bound with NADP and ApoCBR1: CBR1 not bound to any substrates. (B) Apo-NADP: CBR1 bound with NADP. 4Z3D: CBR1 bound with GSH in closed form and NADP. (C) 2PFG: CBR1 bound with BiGF2 and NADP. (D) 3BHM: CBR1 bound with S-hydroxymethylglutathione and NADP. (E) 3BHJ: CBR1 bound with GSH, OH-PP and NADP.
3.5. MD simulation of CBR1 bound with 2,3-butanedione in the presence or absence of GSH
2,3-Butanedione (diacetyl) was used as a model of a low-mw low-affinity ligand for CBR1. MD simulation was run to evaluate the changes in the binding ability of 2,3-butanedione as well as the structural changes in CBR1 in the presence or absence of GSH. GSH was modelled in open or “ON” form to allow docking of 2,3-butanedione in the active site, while its carbonyl group forms hydrogen bonds with the catalytic residues SER139 and TYR193.
Despite the initial localization of 2,3-butanedione at the active site with its carbonyl group facing NADPH and making hydrogen bonds with two residues of the catalytic triad, the compound was unable to persist in this position during the MD simulation. MD simulation of 2,3-butanedione in the absence or presence of GSH was performed. In either case, 2,3-butanedione was released from the active site soon after the execution of the run macro. Coinciding with its low affinity for CBR1, in the absence of GSH, 2,3-butanedione was released from its binding site after 5 ns of simulation. GSH was found to add additional steric constraints against the presence of 2,3-butanedione in CBR1, as the compound was detached from the active site within only the first 2 ns. Supplementary videos 2 and 3 represents MD simulation of 2,3-butanedione in the absence or presence of GSH, respectively.
4. Discussion
MD simulation has been used to reveal structural changes in proteins and enzymes in response to various ligands or protein–protein interactions [Citation25,Citation26]. By utilizing MD technique, structural dynamic changes that cannot be easily handled by other experimental techniques can be evaluated.
4.1. Moderate binding strength between GSH and CBR1
The glycine moiety of GSH did not strongly bind to the active site of the enzyme. This finding agrees with the binding strength of GSH with CBR1 measured by isothermal titration calorimetry. The measured GSH dissociation constant was 25 µM [Citation14]. In terms of binding strength, this value is considered to indicate moderate binding strength. The lack of strong binding between GSH and CBR1 is, therefore, essential to allow the access of highly specific ligands to the active site.
4.2. The contribution of substructure moieties of GSH to the binding interactions with CBR1
There were noticeable differences between the contributions of the glutamate and glycine moieties of GSH to its interaction with CBR1. The overall configuration indicates the generally stronger contribution of glutamate than glycine to GSH binding. The glutamate α-amine group establishes strong hydrogen bonds with the hydroxyl group of GLN105. In addition, its carboxylate group forms strong hydrogen bonds with the amino groups of the ALA192 and TYR193 side chains. In contrast, the glycine moiety, which is pointing toward the active site aperture to NADPH, forms hydrogen bonding interactions with the terminal hydroxyl group and the side chain carboxylates of SER139 and TYR193. This finding underscores the contribution of the glycine moiety to GSH binding due to the general weakness of hydrogen bonds between hydroxyl and carboxylate moieties. In conclusion, the initial binding interaction of GSH substructures confirms the notion that the glutamate moiety is fixed, while the glycine moiety is mobile due to its weak interactions with CBR1.
4.3. Variations of GSH placement site during MD
Variations in GSH configurations were previously identified in two different crystal structures (4Z3D and 3BHJ). The structures showed complete alignment with low RMSD. Interestingly, the glycine moiety of GSH showed marked deviations in its position. Thus, the glycine moiety might behave as an on/off switch to protect NADPH and control the substrates entering the active site [Citation14]. The GSH binding site showed additional interesting features: it acts as an on/off switch with right-left movement capacity. This capability is also regulated by the slipping of the glutamate moiety in the up–down direction (Figure ). This feature presumably allows better adaptation to substrates of variable lengths and sizes.
4.4. GSH affects the specificity of CBR1 toward low-affinity ligands
The low-affinity ligand 2,3-butanedione showed unstable binding to CBR1 with an empty catalytic site: it was released from the site within 10 ns, and during this time, it showed displacements and vibrations within the binding site. In the presence of GSH, the space accessible to 2,3-butanedione within the active site of CBR1 was marginally reduced to the point where the compound was released at an earlier time of only 2 ns. This result confirms the role of GSH in maintaining CBR1 substrate specificity and controlling the type of ligands remaining at the active site.
In order to get deep insights into the compounds recognition by CBR1, the MM/GBSA binding free energy for the binding of two poor substrates and one strong ligand were compared. The Brenda-enzymes website was browsed to retrieve the substrates for the binding energy studies. The Km values were 18, 1 and 0.000053 mM for 2,3-butanediol, phenylglyoxal and 4-benzoylpyridine, respectively. The former two ligands were taken as poor affinity ligands, while 4-benzoylpyridine was used as high affinity ligand. The results of MM/GBSA binding energy was perfectly coinciding with the estimated Km values. The obtained MM/GBSA binding energy values were −31.91, −32.90 and −44.2 for 2,3-butanediol, phenylglyoxal and 4-benzoylpyridine, respectively. These values agrees with the experimentally measured substrates affinity. The decomposition of binding energy indicated the importance of lipophilic interaction, which was positively correlated with the estimated Km values. The high affinity substrate was associated lower coulombic and hydrogen bond scores and higher lipo score (Table ).
Table 2. The MM/GBSA binding energy and energy decomposition analysis.
4.5. Flexibility of CBR1 to adapt to GSH adducts
The presence of several GSH hydrocarbon adducts did not significantly change the overall profile of the structure RMSD during MD simulation (Figure ). While the structures 3BHM and 2PFG contain GSH adducts (BiGF2 and S-hydroxymethylglutathione), there was no significant difference in RMSD compared with that in the presence of only GSH (3BHJ), as shown in Figure . Additionally, the flexible loop ILE92-PHE102 was homogenously changed across all possible ligands, including GSH and its adducts. The lack of changes in this loop in 3BHJ, compared to ApoNADP is due to the weak binding and release of the bound ligand from its binding site upon starting the simulation.
The VAL230-TYR251 loop forms major interactions with NADP and shows marked flexibility during MD. A severe decrease in the flexibility of this loop was observed in the presence of BiGF2 (PDB ID 2PFG, Figure (C)). The initial localization of BiGF2 before simulation reveals hydrogen bonding interactions with MET234. After 100 ns of simulation, an additional interaction with LYS238 was observed, thereby limiting the structural plasticity of this loop. This effect might contribute to the observed weak inhibitory activity of BiGF2 for CBR1 (Ki> 200 µM) [Citation13].
This study highlights the significance of GSH as a molecular switch in CBR1 activity. Further structural studies are recommended to understand its impact on substrates of variable affinities.
5. Conclusions
GSH acts as a molecular switch in the active site of CBR1. The glycine moiety of GSH showed dynamic changes by moving into on and off positions. This action was spontaneous and occurred in the absence of any other molecules. These changes are almost fixed in the presence of a bound substrate. The impact of GSH on the binding of low-affinity substrates highlights its steric effects on a ligand entering the CBR1 active site.
Supplemental Material
Download MP4 Video (880.3 KB)Supplemental Material
Download MP4 Video (1.7 MB)Supplemental Material
Download MP4 Video (15.3 MB)Acknowledgements
The authors acknowledge the Deanship of Scientific Research at King Faisal University for the financial support under Nashir Track (Grant No. 186027).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data is available within the manuscript.
Additional information
Funding
Related Research Data
References
- Pearson PG, Wienkers LC. Handbook of drug metabolism. Boca Raton (FL): CRC Press; 2019.
- Hoffmann F, Maser E. Carbonyl reductases and pluripotent hydroxysteroid dehydrogenases of the short-chain dehydrogenase/reductase superfamily. Drug Metab Rev. 2007;39(1):87–144. doi:10.1080/03602530600969440.
- Matsunaga T, Shintani S, Hara A. Multiplicity of mammalian reductases for xenobiotic carbonyl compounds. Drug Metab Pharmacokinet. 2006;21(1):1–18. doi: 10.2133/dmpk.21.1
- El-Hawari Y, Favia AD, Pilka ES, et al. Analysis of the substrate-binding site of human carbonyl reductases CBR1 and CBR3 by site-directed mutagenesis. Chem Biol Interact. 2009;178(1-3):234–241. doi:10.1016/j.cbi.2008.11.004.
- Hartmanova T, Tambor V, Lenco J, et al. S-nitrosoglutathione covalently modifies cysteine residues of human carbonyl reductase 1 and affects its activity. Chem Biol Interact. 2013;202(1-3):136–145. doi:10.1016/j.cbi.2012.12.011.
- Bohren KM, Wermuth B, Harrison D, et al. Expression, crystallization and preliminary crystallographic analysis of human carbonyl reductase. J Mol Biol. 1994;244(5):659–664. doi:10.1006/jmbi.1994.1762.
- Rotondo R, Moschini R, Renzone G, et al. Human carbonyl reductase 1 as efficient catalyst for the reduction of glutathionylated aldehydes derived from lipid peroxidation. Free Rad Biol Med. 2016;99:323–332. doi:10.1016/j.freeradbiomed.2016.08.015.
- Carlquist M, Frejd T, Gorwa-Grauslund MF. Flavonoids as inhibitors of human carbonyl reductase 1. Chem Biol Interact. 2008;174(2):98–108. doi:10.1016/j.cbi.2008.05.021.
- Wermuth B. Purification and properties of an NADPH-dependent carbonyl reductase from human brain. Relationship to prostaglandin 9-ketoreductase and xenobiotic ketone reductase. J Biol Chem. 1981;256(3):1206–1213.
- Mordente A, Meucci E, Martorana GE, et al. Human heart cytosolic reductases and anthracycline cardiotoxicity. IUBMB Life. 2001;52(1-2):83–88. doi:10.1080/15216540252774829.
- Seliger JM, Martin H-J, Maser E, et al. Potent inhibition of human carbonyl reductase 1 (CBR1) by the prenylated chalconoid xanthohumol and its related prenylflavonoids isoxanthohumol and 8-prenylnaringenin. Chem Biol Interact. 2019;305:156–162. doi: 10.1016/j.cbi.2019.02.031
- Breysse DH, Boone RM, Long CM, et al. Carbonyl reductase 1 plays a significant role in converting doxorubicin to cardiotoxic doxorubicinol in mouse liver, but the majority of the doxorubicinol-forming activity remains unidentified. Drug Metab Dispos. 2020;48(3):187–197. doi: 10.1124/dmd.119.089326
- Bateman R, Rauh D, Shokat KM. Glutathione traps formaldehyde by formation of a bicyclo[4.4.1]undecane adduct. Org Biomol Chem. 2007;5(20):3363–3367. doi:10.1039/b707602a.
- Liang Q, Liu R, Du S, et al. Structural insights on the catalytic site protection of human carbonyl reductase 1 by glutathione. J Struct Biol. 2015;192(1):138–144. doi:10.1016/j.jsb.2015.09.005.
- Pirolli D, Giardina B, Mordente A, et al. Understanding the binding of daunorubicin and doxorubicin to NADPH-dependent cytosolic reductases by computational methods. Eur J Med Chem. 2012;56:145–154. doi:10.1016/j.ejmech.2012.08.023.
- Staab CA, Hartmanova T, El-Hawari Y, et al. Studies on reduction of S-nitrosoglutathione by human carbonyl reductases 1 and 3. Chem Biol Interact. 2011;191(1-3):95–103. doi:10.1016/j.cbi.2011.01.016.
- Bateman R, Rauh D, Shokat KM. Glutathione traps formaldehyde by formation of a bicyclo [4.4. 1] undecane adduct. Org Biomol Chem. 2007;5(20):3363–3367. doi: 10.1039/b707602a
- Pilka ES, Niesen FH, Lee WH, et al. Structural basis for substrate specificity in human monomeric carbonyl reductases. PLoS One. 2009;4(10):e7113), doi:10.1371/journal.pone.0007113.
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430
- Krieger E, Vriend G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics. 2014;30(20):2981–2982. doi: 10.1093/bioinformatics/btu426
- Kandeel M, Al-Taher A, Al-Nazawi M, et al. Substructural dynamics of the phase-I drug metabolizing enzyme, carbonyl reductase 1, in response to various substrate and inhibitor configurations. Trop J Pharm Res. 2019;18(8):1635–1641.
- Krieger E, Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36(13):996–1007. doi:10.1002/jcc.23899.
- Tanaka M, Bateman R, Rauh D, et al. An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol. 2005;3(5):e128. doi:10.1371/journal.pbio.0030128.
- Bateman RL, Rauh D, Tavshanjian B, et al. Human carbonyl reductase 1 is an S-nitrosoglutathione reductase. J Biol Chem. 2008;283(51):35756–35762. doi:10.1074/jbc.M807125200.
- Venugopala KN, Chandrashekharappa S, Pillay M, et al. Synthesis and structural elucidation of novel benzothiazole derivatives as anti-tubercular agents: In-silico screening for possible target identification. Med Chem. 2018. doi: 10.2174/1573406414666180703121815
- Kandeel M, Kitade Y. Molecular dynamics and binding selectivity of nucleotides and polynucleotide substrates with EIF2C2/Ago2 PAZ domain. Int J Biol Macromol. 2018;107(Pt B):2566–2573. doi:10.1016/j.ijbiomac.2017.10.145.