
ABSTRACT
Favipiravir is a broad spectrum antiviral drug that has shown activity against many viruses. Sequel to the recent outbreak of COVID-19, favipiravir is investigated as one of the potential drugs for the treatment of SARS-CoV-2. To augment these efforts, this article reports the rotational isomers, tautomeric states, electronic and spectral properties of favipiravir and its five analogues (Cl, Br, H, CN and CH3) using quantum chemcial code. The enol forms are more stable and the calculated keto–enol relative energies are in the range of 7.86–10.72 kcal/mol in the gas phase and 1.07–3.46 kcal/mol in the solution phase. The relative stabilization of the more polar keto structure in water environment leads to a significant reduction in keto–enol relative energy by 68% for T705, 71% for T705–Cl and T705–Br, 86% for T1105 (H), 88% for T705–CN and 80% for T705–CH3. The density functional theory and time-dependent density functional theory with the 6-311++G(d,p) basis set were used in the computation. The theoretical results were successfully compared with available experimental and theoretical data.
1. Introduction
Favipiravir (C5H4FN3O2, 6-fluoro-3-hydroxy-2-pyrazinecarboxamide, T705) is a pyrazinecarboxamide based antiviral medication that has demonstrated activity against a number of viral infectious diseases such as influenza viruses (types A, B and C) [Citation1], H5N1 virus [Citation2], hepatotropic pheleboviral disease [Citation3], West Nile virus [Citation4], Norwalk virus (norovirus) [Citation5], encephalitis viruses [Citation6], arenaviruses [Citation7], bunyavirus [Citation7], and Ebola virus [Citation8,Citation9]. Thus, favipiravir which is sold under the trade name Avigan is classified as a broad-spectrum antiviral agent that inhibits the replication of ribonucleic acid (RNA) viral infections [Citation10]. Favipiravir was first reported in 2000 by Toyama Chemical Co., Ltd, Japan [Citation11]. Due to its successful antiviral potency, various researchers have made considerable efforts to modify, optimize, and develop facile and cost effective routes to the synthesis of favipiravir [Citation12–15]. Sequel to the recent outbreak of the novel Coronavirus disease 2019 (COVID-19), an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) across the world, favipiravir is investigated as one of the potential drugs for the experimental treatment of the novel coronavirus disease [Citation16–26].
Similarly, 3-hydroxy-2-pyrazinecarboxamide (T1105) which is a structural analogue of T705 has shown a good antiviral activity against influenza virus [Citation27] and foot-and-mouth disease virus [Citation9]. The T1105 and the brominated analogue of favipiravir (6-bromo-3-hydroxy-2-pyrazinecarboxamide, here after designated as T705–Br) are useful precursors for the synthesis of the stable and effective antiviral T1105–ribonucleotide and T705–Br–ribonucleotide prodrugs [Citation28]. The chemical structures of favipiravir and its structural analogues are provided in Figure .
Figure 1. Chemical structures of favipiravir and its structural analogues.
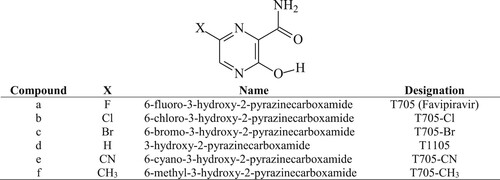
Favipiravir has a rotating carboxamide (CONH2) moiety that makes it a potential pseudo base mimicking both guanine and adenine (as shown in Figure ) similar to ribavirin, a broad spectrum antiviral RNA virus mutagen [Citation29]. The virus mutagenic activity of ribavirin which was ascribed to the rotation of its carboxamide functional group could be applied to favipiravir and T1105. Similarly, the rotation of the hydroxyl (OH) group on pyrazine rings of T705 and T1105 can lead to another rotational conformer. In addition, these molecules have the potential to exhibit keto–enol tautomerism which could be of interest in drug design and development to determine which tautomeric form is bound to a target biomolecule. Despite the important applications of favipiravir, there are no systematic and comparative theoretical studies of the conformational analysis, electronic and spectroscopy properties of favipiravir and its structural analogues. Such studies will contribute to the understanding of the molecular structures, vibrational, and electronic properties of favipiravir that could be used in the development of new pyrazinecarboxamide-based antiviral drugs. Thus, in this study, internal rotations of the carboxamide and hydroxyl groups of T705 and T1105 were examined to determine the different conformations of these molecules along the potential energy profile. The relative stability of the tautomeric states was determined in gas and aqueous solution. This work was extended to other structural analogues of T705 in which the fluorine atom was substituted with chlorine, bromine, cyano and methyl atoms/groups (Figure ). In addition, detailed comparative studies of the structural, electronic and spectroscopic properties of the six molecules were carried out and the results were reported herein. The results and data from such studies could be useful at this moment of urgent need of the development of antiviral drugs for the treatment of novel COVID-19.
Figure 2. Rotational isomers of favipiravir and its structural analogues.
2. Computational method
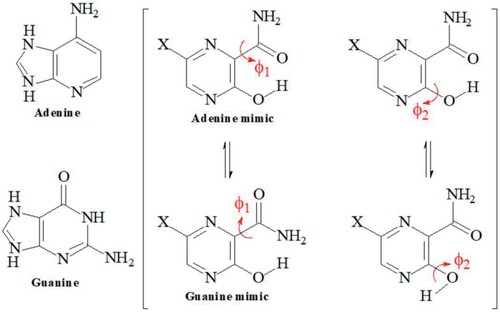
Calculations were done using Density Functional Theory (DFT) method implemented on Gaussian 09 program package [Citation30]. DFT methods are well known to give accurate and reliable description of the molecular geometry, rotational barrier, vibrational frequency and electronic properties of organic compounds [Citation31–36]. The hybrid B3LYP functional [Citation37, Citation38] method and the 6-311++G(d,p) basis set were used for the calculations. The internal rotations along the dihedral angles ϕ1(NC–CO) and ϕ2(NC–OH) (Figure ) were carried out from 0 to 180° to determine the more stable conformer along the energy profiles of favipiravir and its structural analogues (Figure ). The geometry of the minimum energy conformers was optmized with no restrictions, and the optimized structural parameters were used to calculate electronic parameter, NBO atomic charges, IR and UV-visible spectra. The molecules have the potential to exist in keto–enol tautomeric forms. Thus, the geometry optimizations of the tautomeric forms of the studied molecules were carried out to determine the keto–enol relative energies in gas and solution phases. Water was used as a solvent, and the solution calculations were carried out using the Polarizable Continuum Model (PCM), as well as the integral equation formalism variant (IEF-PCM) [Citation39], as implemented in Gaussian 09. Time dependent density functional theory (TD-DFT) was used for the electronic absorption spectra calculations. The normal vibrational modes were characterized on the basis of Potential Energy Distribution (PED) using the VEDA4 program [Citation40]. The PED analysis requires the construction of 3N-6 linearly independent local mode coordinates that represent stretching, bending, and torsional motions in the molecule. The VEDA4 program automatically extracts input data from the Gaussian output files (.log and .fchk, renamed as .fch) and proposes an introductory set of local modes coordinates. More adequate coordinates are then proposed, and optimized by the program to obtain maximal elements of each coordinate of the PED matrix [Citation41].
3. Result and discussion
3.1. Structural parameters and potential energy profile analysis
The structures of favipiravir (T705) and it structural analogues (T705–Cl, T705–Br, T1105, T705–CN and T705–CH3) contain carboxamide (CONH2) and hydroxyl (OH) functional groups attached to the pyrazine at positions 2 and 3 respectively. Gas phase geometry optimization of these molecules was carried out at B3LYP/6-311++G(d,p) without any symmetry constraint and convergence criteria. Figure shows the minimum energy geometry and atom numbering for T705 and its five other structural analogues. The bond lengths and some of the bond angles of the optimized structures are provided in Table along with the experimental values for T705 [Citation13]. The calculated structural parameters for T705 are excellent agreement with the experimental values obtained from the X-ray structure of T705. The root mean square deviation (RMSD) values and the correlation (R2) between the experimental and theoretical values are determined to be 0.084/0.9865 for the bond lengths, and 0.653/0.9852 for the endocylic bond angles of the pyrazine ring.
Figure 3. Minimum energy structures and atom numbering for favipiravir and its analogues.
Table 1. Some of the optimized structural parameters for favipiravir and its analogues.
The carbonyl (C9=O10), hydroxyl (O14−H15) and C6−F8 bond lengths of T705 are calculated to be 1.237, 0.990 and 1.344 Å. These values are in agreement with the experimental values of 1.224, 0.8200 and 1.339 Å. The data in Table show that these molecules adopt a planar geometry in which the pyrazine ring, carboxamide and hydroxyl groups lie in the same plane. The dihedral angles ϕ1(N1–C2–C9–O10) and ϕ2(N4–C3–O14–H15) that connect the carboxamide and hydroxyl groups to the pyrazine ring are calculated to be 180.0° for all the molecules. In general, the geometric parameters of the six molecules are very similar suggesting that the substitution of the fluorine atom of T705 with Cl, Br, H, CN and CH3 has little or no effect on the structural parameter. The pyrazine rings C−N and C−C bond lengths for the six molecules are shorter than the normal C−N single bond value of 1.49 Å [Citation42] and the normal C−C single bond of 1.54 Å [Citation43]. The pyrazine bond lengths and angles are similar to the calculated values reported for 5-chloro-N-(3-nitrophenyl)pyrazine-2-carboxamide [Citation44]. The carbonyl (C9=O10) bond lengths are calculated to be 1.237 for T705, T705–Cl, T705–Br and T705–CN; and 1.238 Å for T1105 and T705–CH3. The hydroxyl (O14−H15) bond lengths are calculated in the range of 0.989–0.994 Å while the N–H bond lengths of the amine group are determined to be 1.007 Å (N11–H12) and 1.009 Å (N11–H12) for all the molecules. All these structural data are in agreement with the theoretical results reported by Lydia et al. [Citation45].
3.2. Potential energy profile analysis
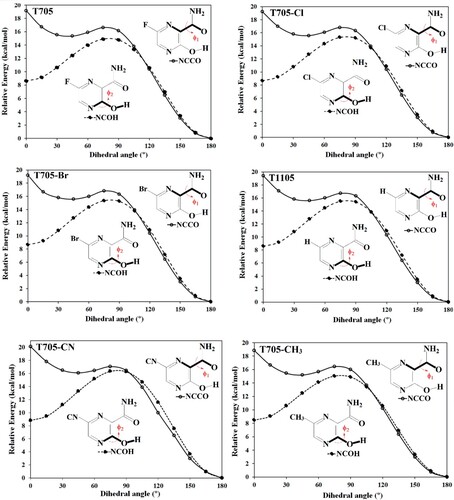
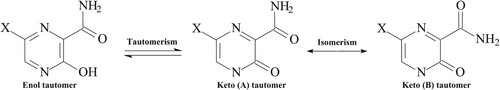
The minimum energy structure of these molecules has the nitrogen (N1) atom of pyrazine ring trans to the oxygen atom (O10) of the carboxamide group (where the N1–C2–C9–O10 dihedral angle (ϕ1) equals to 180.0°). Internal rotation of the carboxamide group from ϕ1 = 0° (where the N1 atom of pyrazine ring is cis to carbonyl O10 atom) to ϕ1 = 180° (where N1 is trans to carbonyl O10) was carried out. For each fixed dihedral angle, all the remaining degrees of freedom in the molecules were optimized. The potential energy scan (PES) for the six molecules is presented in Figure . These molecules have similar PES pattern with one minimum energy structure corresponding to the NCCO trans conformer. It should be noted that the NCCO cis has the highest energy on the energy profile. Thus, the conformer that mimics the guanine structure [Citation46] has the highest energy in the potential energy profile of all the molecules with the trans–cis energy difference of 19.2 kcal/mol for T705 and its haloginated analogues (T705–Cl and T705–Br); 19.4 kcal/mol for T1105, 20.1 kcal/mol for T705–CN, and 18.8 kcal/mol for T705–CH3. Therefore, it could be concluded that the formation of pseudo base guanine-like conformer by this set of molecules is unlikely due to the relatively high energy difference. Similarly, internal rotations of the hydroxyl group from ϕ2 = 0° (where the N4 of the pyrazine ring is cis to the hydroxyl O14) to ϕ2 = 180° (where N4 is trans to hydroxyl O14) were carried out. The molecules show similar PES pattern (Figure ) with the trans NCOH conformer having the lowest energy and the transition state structure at dihedral angle (ϕ2) ≈ 75°.
Figure 4. Potential energy scan for favipiravir and its analogues.
Scheme 1. Keto–enol tautomers and keto rotatioanl isomer for favipiravir and its analogues.
3.3. Keto–enol tautomerism
In addition to the rotational isomers discussed above, these molecules have the potential to exhibit keto–enol tautomerism as shown in Scheme 1. The tautomeric isomers of these molecules could be of interest in drug design and development to determine which tautomeric form is bound to a target biomolecule. Some of the molecules in drug discovery data base are potentially tautomeric, and the knowledge of the exact tautomer in different stages of drug design is very essential. Examples of drugs that have keto–enol potential include milrinone (Primacor), curcumin, warfarin (Coumadin), piroxicam and hydroxyquinoline [Citation47–49]. Thus, the tautomeric isomers of these molecules were optimized at the same level of theory and the keto–enol relative energies (ΔE = Eketo − Eenol) for the six molecules were determined. The keto rotomer (B) was found to be more stable than keto (A) and as such keto (B) forms were used for all tautomeric calculations. The potential energy scan for rotation of the carboxamide group on the keto tautomers of the six molecules are presented in Figure .
Figure 5. Potential energy scan for the keto tautomers of favipiravir and its analogues.
The calculated gas phase keto–enol relative energies are 10.72, 10.34, 10.21, 8.02, 9.23, and 7.86 kcal/mol for T705, T705–Cl, T705–Br, T1105, T705–CN and T705–CH3 respectively. In general, enol form predominates at equilibrium. However, for this set of molecules, the enol form is energetically favoured due to the aromatic character of the pyrazine ring which is absent in the ketone form. In addition, the enol form is stabilized by the intramolecular hydrogen bond between the hydrogen atom (H15) of the hydroxyl group and the oxygen atom (O10) of the carbonyl group. The keto form, however, has two non conjugated carbonyl groups. The gas phase O14−H15 … O10 intramolecular hydrogen bond distances are 1.723 Å (T705), 1.719 Å (T705–Cl), 1.720 Å (T705–Br), 1.715 Å (T1105), 1.697 Å (T705–CN), 1.724 Å (T705–CH3), and the experimental value of 1.880 Å was reported for crystal structure of T705. The dipole moments, keto–enol relative energies and Gibb’s free energies, and the percentage of the enol tautomer in both gas and aqueous media for all the molecules are presented in Table . The results suggest that the keto forms are unlikely to exist in the gas phase.
Table 2. Dipole moment (Debye), keto–enol relative energy (kcal/mol), keto–enol relative free energy (kcal/mol), and percentage enol for favipiravir and its analogues in gas and aqueous medium.
The geometry optimization of the tautomeric forms of the studied molecules was carried out in aqueous medium using the IEF-PCM solvation model [Citation39] which was previously used for organic compounds [Citation47,Citation48]. The results of the calculations of the energies of the keto and enol forms in water enviroment as a solvent show strong stabilization of keto form. Although the aromaticity is not preserved, the keto form predominates in the in aqueous media. This is because the keto forms have higher dipole moment than the enol forms. The solution phase calculated relative energies are 3.46, 3.01, 2.92, 1.12, 1.07, and 1.53 kcal/mol for T705, T705–Cl, T705–Br, T1105, T705–CN and T705–CH3 respectively. The relative stabilization of the more polar keto structures in water enviroment leads to significant reduction in keto–enol energy differences. The reduction in energy gap is about 7 kcal/mol (for T705, T705–Cl, T705–Br, and T1105), 8 kcal/mol (for T705–CN) and 6 kcal/mol (for T705–CH3). The tautomeric N−H proton in keto form is available for interaction with proton acceptor solvent. In enol form, however, the interaction of tautomeric O−H proton with water molecules distrupts the intramolecular hydrogen bond. The effect of water in keto–enol equilibrium shift by the stabilization of one of the tautomers has been observed in piroxicam [Citation47], curcumin [Citation48] and pyrazine carboxamide based drugs [Citation49]. The substitution of fluorine atom in T705 with Cl, Br, H, CN and CH3 leads to further stabilization of the keto tautomers in aqueous medium. The percentage of enol (provided in the last column of Table ) decreases upon the replacement of flourine atom in T705, with T-1105 having the highest reduction. However, the M06-2X/def2TZVP and TautLYP/6-31++G(d,p) showed that the T-1105 is unlikely to exist in the aqueous phase [Citation50] but the gas phase results agree with what is reported herein. Though prediction of tautomeric equilibrium depends on the method of theoretical calculation, the availability of experimental data will help in clarifying the theoretical results.
3.4. Infra-red and UV-visible absorption spectra
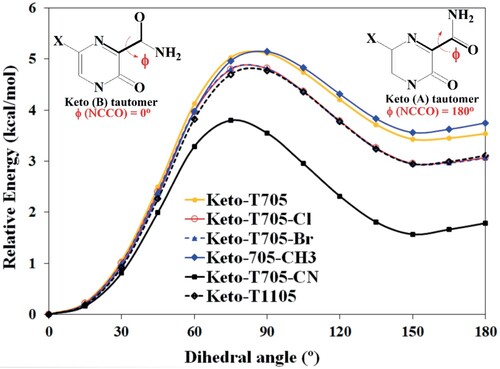
The IR vibrational wavenumbers were obtained from the frequency calculations using the structural parameters of the more stable conformer of the studied molecules. T705, T705–Cl, T705–Br, and T1105 are composed of 15 atoms with 39 active vibrational modes. On the other hand, T705–CN and T705–CH3 contain 16 and 18 atoms which give 42 and 48 vibrational modes respectively. The vibrational wavenumbers (scaled and unscaled) and their corresponding intensities for the six molecules are provided in Tables S1–S6 of the supplementary information along with the proposed assignments of the vibrational modes. These molecules contain amide and hydroxyl functional groups at positions 2 and 3 of the pyrazine ring. For all the molecules, the calculated vibrational wavenumbers at around 3590 and 3560 cm−1 are attributed to N−H asymmetric and symmetric stretching. These modes are pure NH vibrations with 99% contributions to PED. The carbonyl C=O vibrations of these molecules are calculated to be around 1600 cm−1, a wavenumber shorter than normal carbonyl absorption but typical of C=O stretching absorption band (amide I band) due to the resonance effect [Citation51]. The resonance effect in the amide group increases the C=O bond length and reduces the frequency of absorption. The OH stretching modes are calculated to be around 3200 cm−1 (for T705, T705–Cl, T705–Br, T1105 and T705–CH3) and 3145 cm−1 (for T705–CN). These are all pure OH vibrations with 99% contributions to PED.
The pyrazine ring CH stretching modes, which are all 100% pure modes with weak intensities, are estimated to be at 3045 cm−1 for T705–CH3, 3064 cm−1 for T705–CN, and at around 3070 cm−1 for T705, T705–Cl, T705–Br and T1105. The C≡N stretching of T705–CN is calculated to be at 2263 cm−1, while the CH stretching of T705–CH3 is determined to be at 3010, 2987 and 2934 cm−1. The computed vibrational wavenumbers computed in this work agree quite well with those of previous theoretical study [Citation45] as well as those reported for 5-chloro-N-(3-nitrophenyl)pyrazine-2-carboxamide [Citation44]. Figure shows the simulated infrared spectra of all the six studied molecules. This figure reveals the spectra characteristics of the molecules. The peaks associated with the carbonyl, amino, hydroxyl, cyano and methyl groups are assigned in the individual spectrum of the molecules.
Figure 6. Simulated vibrational Infrared spectra of favipiravir and its analogues.
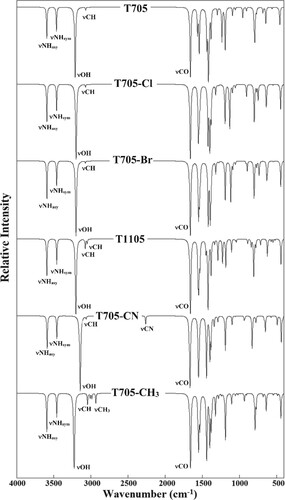
Figure shows the UV-visible absorption spectra of the studied molecules in methanol as a solvent. Time Dependent-DFT (TDDFT) with 6-311++G(d,p) basis set was used for the UV-visible absorption spectral calculations using the equilibrium structural parameters of molecules. The UV-visible spectrum absorption maximum values for T705, T705–Cl, T705–Br, T1105, T705–CN and T705–CH3 were calculated to be 311, 315, 317, 323, 320 and 326 nm. These maximum absorption values are attributed to HOMO-1 → LUMO transition with percentage molecular orbital contribution of about 99%. For the halogenated analogues, the aborption maximum (λmax) slightly shifts to longer wavelength as the size of the halogen atom increases. The absorption wavelength (λ), excitation energies (E), and oscillator strengths (f) obtained from the TDDFT calculations for the first six transition of the studied molecules are provided in Table S7 of the supporting information. For all the molecules, the HOMO → LUMO transitions have the highest oscillator strength, and thus, the most probable transition. This transition is the second transition for all the molecules (Table S7).
Figure 7. Simulated UV spectra of favipiravir and its analogues at TD-DFT/B3LYP/6-311++G(d,p).
3.5. Electronic properties
Figure shows the 3D plots of Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) for T705 and its structural analogues. These plots are generated from optimized and frequency calculations. As clearly observed in the figure, the HOMO orbitals are spread over the entire molecules, while the LUMO orbitals are localized on the pyrazine ring and carboxamide group. The similarities in HOMO and LUMO energy values have reflected in the HOMO–LUMO energy gaps which are determined to be 4.52 (T705), 4.52 (T705–Cl), 4.52 (T705–Br), 4.52 (T1105), 4.52 (T705–CN) and 4.52 eV (T705–CH3). For the halogenated molecules, the substitution of fluorine atom of T705 with chlorine (T705–Cl) and bromine (T705–Br) atoms leads to a slight decrease in HOMO–LUMO energy gap (T705(F) < T705–Cl < T705–Br) as the size of the halogen atom increases. Among the six molecules, T1105 has the highest HOMO–LUMO energy gap, while T705–Br has the lowest. The calculated HOMO, LUMO and HOMO–LUMO energy values are presented in Table along with the global reactivity descriptors including ionization potentials (IP), electron affinity (EA), electronegativity (χ), chemical hardness (η), chemical potential (μ), chemical softness (S) and electrophilicity index (ω). For halogenated molecules, there is no trend in the relationship between the electronegativity of the halogen atoms (F, Cl, and Br) and the electron affinity of the compounds (T705, T705–Cl and T705–Br). However, the substitution of fluorine atom with less electronegative Cl and Br atoms slightly decreases the IP, EN, η and ω of the molecules. As evident in Table , T705–CN has the highest IP (7.81 eV), EA (3.13 eV), EN (5.47 eV) and μ (−5.47 eV); T705 has the highest χ (5.77 eV); T705–Br has the highest S (0.23 eV−1); and T1105 has the highest η (5.47 eV).
Figure 8. Frontier molecular orbitals for favipiravir and its analogues.
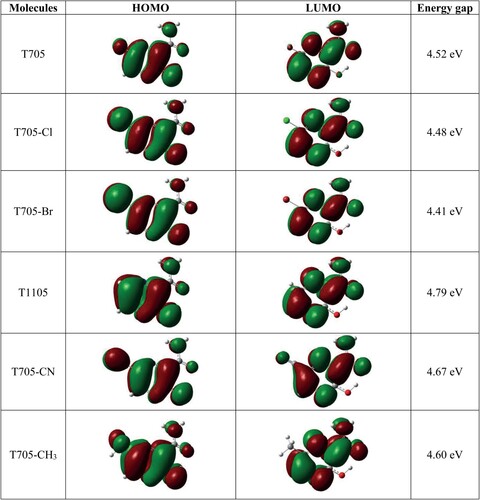
Table 3. Calculated HOMO and LUMO energies, HOMO–LUMO energy gap, and global reactivity descriptors for favipiravir and its analogues.
3.6. Atomic charges and molecular electrostatic potentials
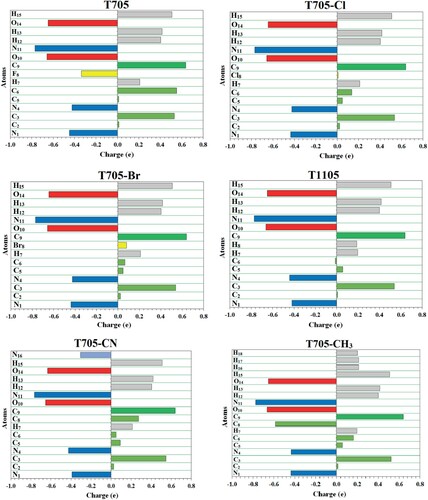
The atomic charges were calculated from the B3LYP/6-311++G(d,p) equilibrium geometrical structures of the molecules using NBO method of the Gaussian 09. The atomic charges of T705 along with five other structural analogues are illustrated in Figure , and the corresponding values are given in Table S8. The charge distribution for all the molecules is similar but the magnitude of the charges is slightly different. For all the molecules, the N1, N4, O10, N11 and O14 are negatively charged due to a relatively higher electronegativity of these atoms than carbon and hydrogen atoms. The hydrogen atoms (H7 and H15) are positively charged but hydroxyl H15 is more positive due to the electron withdrawing effect of adjacent O14. The hydrogen atoms (H12 and H13) and carbonyl carbon atom C9 of the carboxamide group are all positively charged with almost equal magnitude for all the molecules. The C2, C3 and C5 of the pyrazine ring are positive but C3 is more positive due to the influence of the hydroxyl O14 attached to it. The significant difference in the charge distributions of the studied molecules is the atomic charge of C6 which is calculated to be 0.552e for T705, 0.136e for T705–Cl, 0.067e for T705–Br, −0.012e for T1105, and 0.050e for T705–CN and 0.165e for T705–CH3.
Figure 9. NBO atomic charges for favipiravir and its analogues.
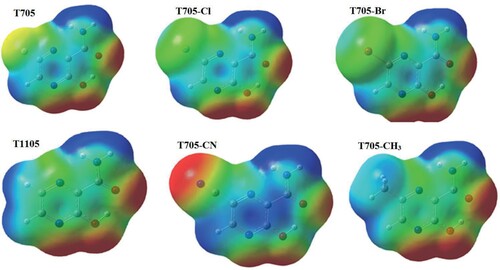
The possible interaction sites of the studied molecules are illustrated by molecular electrostatic potentials (MEP) surfaces (Figure ). The MEP surface is colour coded to depict the variation of charge density. The regions that are prone to nucleophilic and electrophilic attacks are coded blue (positively charged) and red (negatively charged) respectively. The charge density on MEP surfaces are in consistent with the charge distribution provided in Table S8 and Figure . For all the molecules, the electrophilic region lies around amine moiety of carboxamide group. In contrast, the nucleophilic region spreads over the carbonyl oxygen, hydroxyl oxygen and the pyrazine ring nitrogen (N1) ortho to the hydroxyl group. In addition, T705–CN has a strong nucleophilic centre around the cyano group. The nucleophilic nitrogen atom N1 represents the potential protonation site for these molecules especially in biologically active environment. The pyrazine represents the potential site for intramolecular interaction. T1105 and T705–CH3 have a potential electrophilic region around methyl and hydrogen moieties attached to C5 and C6.
Figure 10. Molecular electrostatic potential energy surface (MEP) of favipiravir and its analogues.
4. Conclusion
Theoretical analyses of a broad-spectrum antiviral drugs favipiravir (T705) and 3-hydroxy-2-pyrazinecarboxamide (T1105) and their chlorine, bromine, cyano and methyl anologs were systematically carried to determine the more stable rotational isomer and tautomeric form in gas and aqueous phase. The results show that the formation of pseudo base guanine-like rotational conformer by this set of molecules is unlikely due to the relatively high energy differences. The more stable conformers have both dihedral angles ϕ1 (N1–C2–C9–O10) and ϕ2 (N4–C3–O14–H15), which are equal to 180.0°. This is an orientation that favours intramolecular hydrogen bonding. For the tautomeric studies, the aromatic enol tautomer is energetically more stable in the gas phase and aqueous medium. However, the tautomeric equilibrium is shifted towards the keto form in aqueous because the water environment stabilizes the more polar keto tautomer. The results suggest that the keto forms of the studied molecules are unlikely to exist in the gas phase, while the solution phases are predominantly enol tautomers with traces of keto tautomer except for T1105 with about 25% of keto form. The six molecules exhibit similar electronic properties, molecular electrostatic potential and charge distributions, and are therefore expected to have similar characteristics. This research gives precise and invaluable information that will help in understanding the structures and properties of favipiravir that could be used in the development of new pyrazinecarboxamide-based antiviral drugs.
Supplemental Material
Download MS Word (52.7 KB)Acknowledgements
The author is grateful to Mohammed Awwal Saidu of Jubail English Language and Preparatory Year Institute for proofreading the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Notes
a The atom numbering is given in Figure .
bExperimental values for T705 compounds taken from reference [Citation13].
a Keto–enol relative energy, ΔE = Eketo − Eenol.
bKeto–enol relative free energy, ΔG = Gketo − Genol.
cThe percentage of enol at 298.15 K is calculated using Keq derived from the equation ΔG = RT ln Keq.
a |ΔE| = EHOMO − ELUMO, I = −EHOMO, A = −ELUMO, χ = (I + A)/2, η = (I − A)/2, μ = −(I + A)/2, S = 1/(2η), and ω = μ2/2η.
References
- Furuta Y, Takahashi K, Fukuda Y, et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother. 2002;46:977–981.
- Kiso M, Takahashi K, Sakai-Tagawa Y, et al. T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc Natl Acad Sci USA. 2010;107:882–887.
- Gowena BB, Wonga M-H, Junga K-H, et al. Efficacy of favipiravir (T-705) and T-1106 pyrazine derivatives in phlebovirus disease models. Antiviral Res. 2010;86:121–127.
- Morrey JD, Taro BS, Siddharthan V, et al. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res. 2008;80:377–379.
- Rocha-Pereira J, Jochmans D, Dallmeier K, et al. Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem Biophys Res Commun. 2012;424:777–780.
- Yamada K, Noguchi K, Komeno T, et al. Efficacy of favipiravir (T-705) in rabies postexposure prophylaxis. J Infect Dis. 2016;213:1253–1261.
- Gowen BB, Wong MH, Jung KH, et al. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother. 2007;51:3168–3176.
- Oestereich L, Lüdtke A, Wurr S, et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 2014;105:17–21.
- Furuta Y, Takahashi K, Shiraki K, et al. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009;82:95–102.
- Zhang T, Zhai M, Ji J, et al. Recent progress on the treatment of Ebola virus disease with favipiravir and other related strategies. Bioorg Med Chem Lett. 2017;27:2364–2368.
- Furuta Y, Takahashi K, inventors; Toyama Chemical Co., Ltd., assignee. Nitrogenous heterocyclic carboxamide derivatives or salts thereof and antiviral agents containing both. CA2339272A1. 2000 Mar 2.
- Zhang T, Kong L, Li Z, et al. Synthesis of favipiravir. Chin J Pharm. 2013;44:841–844.
- Shi F, Zongtao L, Kong L, et al. Synthesis and crystal structure of 6-fluoro-3-hydroxypyrazine-2-carboxamide. Drug Discov Ther. 2014;8:117–120.
- Wang W, Liu M, Xiao X, et al. Synthesis of favipiravir. J Int Pharm Res. 2015;42:220–224.
- Liu F-L, Li C-Q, Xiang H-Y, et al. A practical and step-economic route to favipiravir. Chem Pap. 2017;71:2153–2158.
- Chen C, Huang J, Cheng Z, et al. Favipiravir versus arbidol for COVID-19: a randomized clinical trial; medRxiv; preprint; infectious diseases (except HIV/AIDS); 2020.
- Dong L, Hu S, Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov Ther. 2020;14(1):58–60.
- Agrawal U, Raju R, Udwadia ZF. Favipiravir: A new and emerging antiviral option in COVID-19. Med J Armed Forces India. 2020;76(4):370–376. DOI:10.1016/j.mjafi.2020.08.004.
- Du Y, Chen X. Favipiravir: pharmacokinetics and concerns about clinical trials for 2019-nCoV infection. Clin Pharmacol Ther. 2020. DOI:10.1002/cpt.1844
- McKee DL, Sternberg A, Stange U, et al. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res. 2020;157:104859.
- Zhang J, Xie B, Hashimoto K. Current status of potential therapeutic candidates for the COVID-19 crisis. Brain Behav Immun. 2020. DOI:10.1016/j.bbi.2020.04.046
- Zhang Y, Xu Q, Sun Z, et al. Current targeted therapeutics against COVID-19: based on first-line experience in China. Pharmacol Res. 2020;157:104854.
- Liu C, Zhou Q, Li Y, et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent Sci. 2020;6(3):315–331.
- Khambholja K, Asudani D. Potential repurposing of favipiravir in COVID-19 outbreak based on current evidence. Travel Med Infect Dis. 2020:101710. DOI:10.1016/j.tmaid.2020.101710
- Jean S-S, Lee P-I, Hsueh P-R. Treatment options for COVID-19: the reality and challenges. J Microbiol Immunol Infect. 2020. DOI:10.1016/j.jmii.2020.03.034
- Ali I, Alharbi OML. COVID-19: disease, management, treatment, and social impact. Sci Total Environ. 2020;728:138861. DOI:10.1016/j.scitotenv.2020.138861
- Huchting J, Vanderlindena E, Van Berwaera R, et al. Cell line-dependent activation and antiviral activity of T-1105, the non-fluorinated analogue of T-705 (favipiravir). Antiviral Res. 2019;167:1–5.
- Huchting J, Vanderlinden E, Winkler M, et al. Prodrugs of the phosphoribosylated forms of hydroxypyrazine carboxamide pseudobase T-705 and its de-fluoro analogue T-1105 as potent influenza virus inhibitors. J Med Chem. 2018;61(14):6193–6210.
- Crotty S, Maag D, Arnold JJ, et al. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6:1375–1379.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A.02. Wallingford (CT): Gaussian, Inc.; 2009.
- Umar Y, Abdulla S, Manirul Haque SK, et al. Theoretical investigation of the molecular structure, vibrational spectra and molecular docking of tramadol using density functional theory. J Chin Chem Soc. 2020;67:62–73.
- Umar Y, Morsy MA. Ab initio and DFT studies of the molecular structures and vibrational spectra of succinonitrile. Spectrochim Acta A. 2007;66(4–5):1133–1140.
- Umar Y, Tijani J, Abdalla S. Conformational Stabilities, Rotational Barriers and Vibrational Spectra of 2- Pyrrolecarboxaldehydes and 3- Pyrrolecarboxaldehydes calculated using Density Functional Theory. J Struct Chem. 2019;60(2):186–197.
- Umar Y, Abdalla S. DFT study of the molecular structure, conformational preference, HOMO, LUMO, and vibrational analysis of 2-, and 3-furoyl chloride. J Solution Chem. 2017;4:741–758.
- Umar Y, Tijani J, Abdalla S. Density functional theory studies of conformational stabilities and rotational barriers of 2- and 3-thiophenecarboxaldehydes. J Struct Chem. 2016;57(8):1543–1553.
- Yeoh PH, Lim KZ, Tan EL, et al. Internal rotation of 2-, 3- and 4-pyridine carboxaldehydes and their chalcogen analogues (S and Se) in the gas and solution phases: a theoretical investigation. J Solution Chem. 2016;45(8):1195–1212.
- Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5653.
- Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789.
- Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem Rev. 2005;105(8):2999–3094.
- Jamroz MH. Vibrational energy distribution analysis: VEDA 4 Program Warsaw; 2004.
- Abdalla S, Umar Y, Mokhtar I. Conformational and vibrational analysis of 2-, 3- and 4-pyridinecarbonyl chloride using DFT. Zeitschrift für Physikalische Chemie. 2016;230(5–7):867–882.
- Lukose J, Panicker CY, Nayak PS, et al. Synthesis, structural and vibrational investigation on 2-phenyl-N-(pyrazin-2-yl) acetamide combining XRD diffraction, FT-IR and NMR spectroscopies with DFT calculations. Spectrochim Acta A Mol Biomol Spectrosc. 2015;135:608–616.
- Chis V, Piranau A, Jurca T, et al. Experimental and DFT study of pyrazinamide. Chem Phys. 2005;316(1–3):153–163.
- Rosline Sebastian SH, Al-Alshaikh MA, El-Emam AA, et al. Spectroscopic, quantum chemical studies, Fukui functions, in vitro antiviral activity and molecular docking of 5-chloro-N-(3-nitrophenyl)pyrazine-2-carboxamide. J Mol Struct. 2016;1119:188–199.
- Rhyman L, Tursun M, Abdallah HH, et al. Theoretical investigation of the derivatives of favipiravir (T-705) as potential drugs for Ebola virus. Phys Sci Rev. 2018;3(9):20170198.
- Naesens L, Guddat LW, Keough DT, et al. Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir). Mol Pharmacol. 2013;84(4):615–629. DOI:10.1124/mol.113.087247.
- Martin YC. Experimental and PKa prediction aspects of tautomerism of drug-like molecules. Drug Discov Today Technol. 2018;27:59–64.
- Ivanova D, Deneva V, Nedeltcheva D, et al. Tautomeric transformations of piroxicam in solution: a combined experimental and theoretical study. RSC Adv. 2015;5(40):31852–31860.
- Manolova Y, Deneva V, Antonov L, et al. The effect of the water on the curcumin tautomerism: a quantitative approach. Spectrochim Acta A. 2014;132:815–820.
- Antonov L. Favipiravir tautomerism: a short theoretical report. ChemRxiv; 2020. DOI:10.26434/chemrxiv.12115620.v1
- Umar Y, Abu-Thabit N, Ramasami P. Experimental FTIR and theoretical investigation of the molecular structure and vibrational spectra of acetanilide using DFT and dispersion correction to DFT. J Theor Comp Chem. 2019;18(2):1950009.