
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The variant double perovskites are considered novel materials for solar cells and optoelectronic applications. Here, we explored electronic, mechanical, and optical characteristics of Cs2SeX6 (X = Cl, Br, I) by density functional theory (DFT) analysis. The tolerance factor between 0.97–1.0 signifies the structural stability of the investigated compounds while thermodynamic and mechanical stabilities were ensured by positive frequencies of phonon dispersion as well as elastic constants. Moreover, the Poisson and Pugh's ratios are explored for brittle and ductile behavior. The Debye and melting temperatures have also been reported through mechanical analysis. The tuning of the bandgap takes place from 3.10 to 2.64 eV and then 1.15 eV by substitution of Cl with Br and I, respectively. The optical spectra show a shift in absorption region from ultraviolet-to-visible. In addition, the low light reflection and optical energy loss range (0.0–3.0 eV) promises potential of the studied potential for solar cells and optoelectronics uses.
1. Introduction
To fulfil the stimulating demand for renewable energy in limited resources is very challenging. The conversion of electrical energy from heat or light energy sources is an essential requirement for renewable energy devices. The efficiency of such devices mainly depends on the selection of particular materials. Perovskite materials are intensively studied by researchers because of their potential applications [Citation1–6]. From this versatile family of compounds, lead-halide-based solar cells have the highest efficiency reported to date [Citation7]. A large number of configurations have been scrutinized of organic-based perovskites from which the formamidine lead triiodide (FAPbI3) is found outstanding. Its power conversion efficiency (PCE) reaches 25.06% which is most suitable for solar cells [Citation8]. Regardless of this achievement, Pb-based materials are not environmentally friendly. The replacement of Pb-based optoelectronic material with other non-hazardous material is still needed extensive research [Citation9,Citation10]. To fill this gap, a number of researchers have reported Pb-free single and double perovskite halides (DPH’s) [Citation11]. In the case of simple perovskites, Parrey et al. [Citation12] performed DFT calculations on RbGeI3 and studied electro-optical properties. In another study on halides, Zhang et al. [Citation13] investigated the electronic properties for CsGeX3 (X = Cl, Br and I) and suggested optoelectronic applications of these compounds. Another strong candidate that emerged in the list of photovoltaic applications is vacancy-ordered double perovskite Cs2SnI6 reported recently [Citation14]. The structure of vacancy-ordered double perovskites is A2XY6, where A is tetravalent and X octavalent cations, while Y is halides (Cl, Br or I). Exploring the hidden attributes of double perovskites useful for optoelectronic applications in industry, Wang et al. [Citation15] have investigated a number of novel DPH’s and suggested possible solar cell applications. Among these materials, Sn- and Ge-based DPH’s are better alternatives of Pb-free materials in novel optical devices because they belong to the same group of Pb [Citation16]. Experimentally Cs-based DPH’s were first prepared by Bagnall et al. [Citation17]. In another study by Morss et al. [Citation18], new Cs-based DPH’s were fabricated. Since the last decade, DPH’s have earned the name in the list of Pb-free compounds. Recently, Cs2AgBiCl6 and Cs2AgBiBr6 are cubic compounds reported that have shown remarkable optical applications in the visible range [Citation19,Citation20]. Furthermore, recently, the defective double perovskites Cs2SeCl6 and Cs2TeCl6 are elaborated for thermoelectric properties based on classical transport theory by BoltzTraP code. The computed lattice constants (10.18 and 10.51Å), formation energies (−23.56 and −24.13 eV), bandgaps (2.95 and 3.10 eV) and tolerance factor (0.728 and 0.725) are reported for Cs2SeCl6 and Cs2TeCl6, respectively [Citation21].
The purpose of this study is to explore hidden traits of Cesium-based DPH’s for solar cell and thermoelectric applications. In this study, we have particularly focused on Cs2SeX6 (X = Cl, Br and I) DPH’s. Based on comprehensive literature survey, the electronic, elastic and optical properties of Cs2SeX6 (X = Cl, Br and I) DPH’s are scarcely being reported to date. We have used a state-of-the-art theoretical approach based on DFT to explore the aforementioned properties.
2. Method for calculations
The structural, electronic, elastic and optical properties of DPH’s were analysed by DFT based on the FP-LAPW method [Citation22] performed in WIEN2k code [Citation23]. Additionally, the exchange–correlation potential used in our study is Pardew Burke Ernzerhof Generalized Gradient Approximation (PBE-GGA) [Citation24]. The PBE-GGA approximation solves the properties in the ground state with high precision; however, the calculated bandgap is underestimated. To circumvent this issue, the bandgap is improved by implementing modified Becke and Johnson Potential of Trans and Balaha (TB-mBJ) [Citation25]. It improves the bandgap very accurately and the most versatile potential calculated through a mathematical equation.
(1)
(1)
where c is convergence factor,
indicates density of states and
denotes its kinetic energy. The charge convergence factor is calculated as
(2)
(2)
where α and β are numeric constant and adjusted in WIEN2k. The number of K-points selected to achieve full convergence was set at 2000. This helped to generate a denser k-mesh of 12 × 12 × 12 k-mesh points. The maximum angular momentum value lmax was adjusted at 7. The RMT × Kmax was set to 8. Where RMT and Kmax are muffin-tin radii and cut-off wave vectors. Maximum Fourier transformation vector Gmax was adjusted to 12. The self-consistent field (SCF) convergence was minimized up to 0.00001 Ry. The ground state optimizations of structural reforms were solved using the Murnaghan equation of state. Further, for phonon calculations, the density functional theory perturbation has been incorporated in the phonopy software to extract the phonon dispersion band structures [Citation26].
3. Results and discussion
In this section, we have reported the structural, electronic, elastic and optical properties of DPH’s in detail. In Section 3.1, structural stability is explained under the Goldschmidt tolerance factor, ground state optimization and enthalpy of formation. Moreover, electronic properties are discussed which includes DOS and bandgap analysis. In Section 3.2, mechanical attributes of the studied compounds is being explained in detail. In the last section, optical properties are discussed to explore possible optical applications.
3.1. Structural reforms and electronic attributes
To calculate the DPH’s structural stability, ground state optimization of the compound is essential. In our study, we have used the Murnaghan equation of states to achieve equilibrium in structure. The stability in structure is addressed by using tolerance factor Tf of cubic crystal systems suggested by Goldschmidt [Citation27].
(3)
(3)
The Tf value around unity portrays cubic structure is stable. In our study, all compounds are stable in the cubic phase. The enthalpy of formation Hf of BPH’s is also calculated using
(4)
(4)
All calculated values are summarized in . The negative value of Hf () ensures the thermodynamic stability of studied materials. Among the studied compounds, Cs2SeCl6 is more stable due to the large enthalpy of formation [Citation28].
Figure 1. (Left) Ground state structural optimizations (a) Cs2SeCl6, (b) Cs2SeBr6, (c) Cs2SeI6. (Right) Crystalline structure of Cs2SeX6 (X = Cl, Br and I).
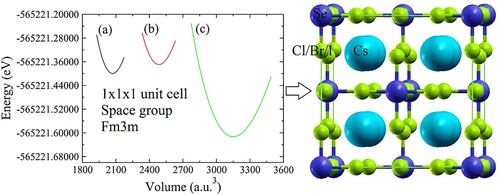
Table 1. Various calculated parameters, for instance, lattice constant ao (Å), bulk modulus Bo (GPa), formation energy Hf (eV), electronic bandgap Eg (eV) and tolerance factor (Tf) of Cs2SeX6 (X = Cl, Br, I).
The density functional perturbation theory (DFPT) has been incorporated to confirm the thermodynamic behaviour of the studied compounds. The computed dispersion plots are illustrated in Figure 2(a–c) which shows the studied compounds have positive frequencies. Therefore, the positive frequencies indicate the studied compounds are dynamically stable. Three acoustic modes of phonons dispersion are accurately zero at Γ symmetry direction which is also the confirmation of dynamical stability [Citation29,Citation30].
Figure 2. The phonon dispersion plots of (a) Cs2SeCl6, (b) Cs2SeBr6, (c) Cs2SeI6, respectively.
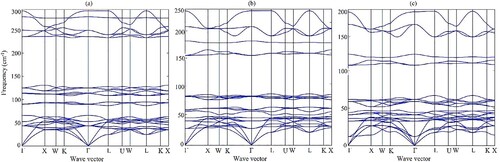
First, the structural, mechanical and thermodynamic stabilities are calculated according to their formalism as shown in the manuscript. However, we relate structural stability with mechanical and thermodynamic stabilities. The structural stability has been calculated from the tolerance factor which depends upon the atomic radii. Mechanical stability has been depicted from born mechanical stability criteria C11 < B < C12, C11–C12 > 0 and C44 > 0 [Citation31]. Therefore, the bulk modulus is volumetric deformation after the applied stress and its value related to the lattice constant. The optimized lattice constant for stable structure and relaxed positions adjusts the bulk modulus to keep positive elastic constants for mechanical stability. Secondly, stable and relaxed structures released more energy which is measured in terms of negative formation energy. Therefore, stable structure measures are directly ensured from formation energy. Thirdly, the acoustic phonons have zero frequencies at Γ and L symmetries as depicted in the dispersion plot in Figure 2(a–c) which is confirmation of structural stability from the thermodynamic approach.
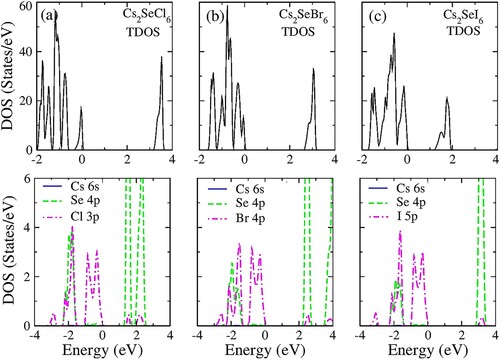
The electronic band structure of DPH’s is depicted in . In this study, spin-polarized TB-mBJ potential is used to investigate the correlation effects. From , all compounds have an indirect bandgap. The bandgap values are depicted in . It is observed that Cs2SeI6 has the smallest bandgap. The nature of the bandgap is further elaborated by the density of states (DOS) plot as shown in . The valence band maxima are contributed mainly by the p orbital of the halogen atom in all compounds. While conduction band minima are formed by Se-p orbital. It can be inferred from the partial DOS that inter-band transition will occur only due to the involvement of Se-p to X-p orbitals.
Figure 3. Electronic band structure of (a) Cs2SeCl6, (b) Cs2SeBr6, (c) Cs2SeI6.
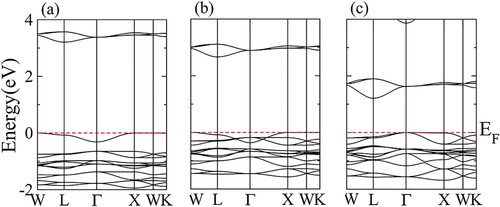
Figure 4. Total DOS and partial DOS of (a) Cs2SeCl6, (b) Cs2SeBr6, (c) Cs2SeI6.
3.2. Mechanical properties
The three elastic constants for the cubic crystals are prescribed as C11, C12 and C44. These constants are calculated using the Charpin method embodied within the WIEN2k code. The stability criteria that cubic compound must satisfy are (C11 > 0), (C44 > 0), (C11 − C12 > 0), (C11 + 2C12 > 0) and (C11 > B > C12) [Citation31]. From , all compounds satisfy the stability criteria. Using these elastic constants, we have calculated bulk modulus (B), Young’s modulus (E) and shear modulus (G) by formulas presented in Muhammed et al. [Citation4]. It is found that Cs2SeCl6 has the highest B, G and E values which suggest that it is more rigid than Cs2SeBr6 and Cs2SeI6. To check whether the compound is ductile or brittle, the Pugh’s (B/G) criteria were used. If the value of B/G > 1.75, the studied material is considered ductile in nature; however, brittle otherwise. In our study, all compounds are found ductile. Additionally, its nature was also confirmed by Poisson’s ratio “υ” for which υ > 0.26 suggests the materials are ductile in nature. Moving further, the Kleinman parameter (ξ) [Citation32] is used to estimate the bond strength whether it is bending or stretching. For ξ near 0, it suggests bond bending while for ξ around unity implies bond stretching. It is observed that Cs2SeI6 has the highest bond-stretching nature. To check the covalent and ionic nature of the bonds present in the unit cell Cauchy parameter is computed as
. The
portrays the dominancy of majority bonds as covalent. While for
ionic-bonding dominancy is expected. From analysis, all studied compounds exhibit positive value which reflects the dominancy of ionic bonding. It is found that Cs2SeCl6 has strong ionic nature than Cs2SeBr6 and Cs2SeI6. We have also checked the Debye temperature to estimate the influence of lattice stability in bonds among atoms by relation [Citation33].
(5)
(5)
where h and k are Plank’s and Boltzmann’s constant, ρ density of alloy, M molecular weight, NA Avogadro number and n number of atoms. Also, V is total velocity computed by relation,
(6)
(6)
where the longitudinal mode of velocity and transverse velocity is given by
(7)
(7)
Ionic compounds exhibit high Debye and melting temperature which is affirmed by results and mentioned in . Melting temperature Tm [Citation34] is estimated by relation
(8)
(8)
Thermal conductivity is used to understand the material response under influence of external heat. The minimum thermal conductivity is estimated by utilizing the Cahill criteria [Citation35,Citation36].
(9)
(9)
where no is no of atoms per unit volume. The highest value of Kmin is observed in the case of Cs2SeCl6 which suggests the material applications in high-temperature sensors. The hardness parameter Ha [Citation37–40] is used to explain the property of a material to resist being dented under stress. It is calculated using the following relation,
(10)
(10)
All computed values are enlisted in which suggests that Cs2SeCl6 has the highest ability to resist the applied stress.
Table 2. The calculated elastic constants (C11, C12 and C44), bulk modulus B (GPa), shear modulus G (GPa), Young modulus E (GPa), Pugh’s ratio (B/G), Poisson ratio (υ), anisotropy factor (A), Kienman parameter (ξ), Lams constant (μ and λ), sound velocities Vℓ, Vt and V (Km/s), Debye temperature D (K), melting temperature Tm (K), hardness Ha (GPa) and minimum thermal conductivity Kmin (Wm−1K−1) of Cs2SeX6 (X = Cl, Br, I).
Table 3. The calculated bandgap Eg (eV), static dielectric constant ε1 (0), static refractive index n (0) and reflectivity R (0) of Cs2SeX6 (X = Cl, Br, I).
3.3. Optical properties
The optical behaviour of materials is strictly dependent on the interaction among light energy and matter, the transition rate of electrons from the valence-to-conduction bands, and the recombination rate. In the case of wide bandgap semiconductors, inter-band transitions are taken into consideration and intra-band transitions are ignored because excitation and recombination within the band are not possible [Citation41]. We have analysed the optical spectra of Cs2SeX6 (X = Cl, Br, I) through graphical representation in Figures and . The dielectric response has been explained by complex dielectric constant ε(ω) = Re ε(ω) + Im ε(ω), where Re ε(ω) is the real part of dielectric constant which enlighten the polarization and dispersion of light energy from lattice while its imaginary part Im ε(ω) describe the absorption of light energy when the frequency of light is greater than the threshold limit. The dependence of Re ε(ω) and Im ε(ω) is explained by the Kramer–Krong relation [Citation42].
(11)
(11)
(12)
(12)
Figure 5. (a) Real part of dielectric constant ε1 (ω), (b) imaginary part of dielectric constant ε2 (ω), (c) absorption coefficient α (ω) and (d) refractive index n (ω) of Cs2SeX6 (X = Cl, Br, I).
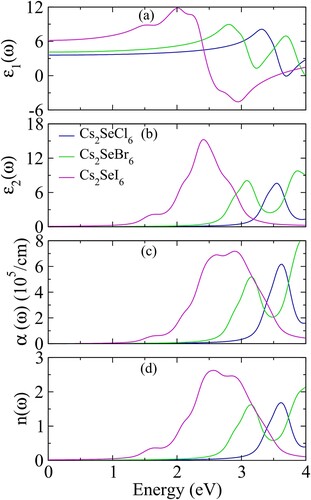
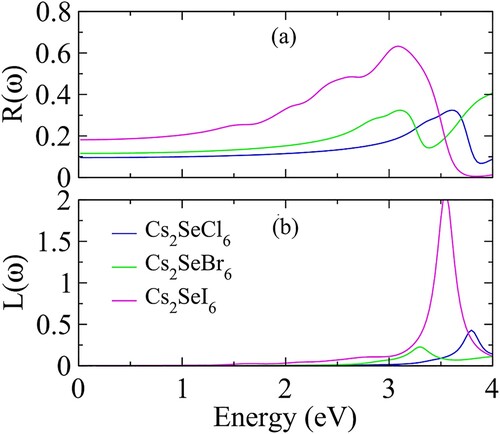
Figure 6. (a) Reflectivity R (ω), (b) optical loss factor L (ω) of Cs2SeX6 (X = Cl, Br, I).
The real dielectric constant Re ε(ω) elaborates the dispersion and polarization of light on interaction with the material of a slightly changing refractive index. The frequency of light depends upon phase velocity which reacts to maximum dispersion and polarized light at plasma resonance of lattice waves. The reported values of Re ε(ω) have been depicted in (a). The zero-energy value static Re ε(0) of Cs2SeCl6 is 3.59 which increases to 4.13 and 6.17 by the replacement of Cl with Br and I because of decreasing bandgap values and large dispersion from Cl to I. The resonance frequencies occur at 3.3 eV (7.9), 2.80 eV (8.93) and 2.0 eV (11.7) for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6, respectively. These are the peak values at which dispersion of light is maximum. Above the resonance frequency, the Re ε(ω) decreases to minimum at 3.69 eV (−0.22), 3.24 eV (1.12) and 2.97 eV (−4.84) for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6, respectively. The two negative peaks for Cs2SeCl6 and Cs2SeI6 show the metallic behaviour for the light which reflects the light falling on the material in this region. Moreover, bandgap Eg and Re ε(0) are according to Penn’s model relation ε(0) ≈ 1 + (ħωp/Eg)2 [Citation43].
The imaginary dielectric constant Im ε(ω) which measures the absorption of light energy is presented in (b). The threshold values of absorption are noted as 3.1, 2.66 and 1.15 eV for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6, respectively [Citation44]. The maximum absorption peaks occur at 3. 54 eV (7.4), 3.09 eV (7.6) and 2.4 eV (15) for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6, respectively which shows the absorption intensity of light increases from Cl to I and ultraviolet region is shifted to visible. Furthermore, absorption bandwidths 0.62 eV (3.2–3.8 eV), 0.75 eV (2.65–3.4 eV) and 1.36 eV (1.78–3.14 eV) increase from Cl to I which depict the Cs2SeI6 is optimal for absorption of light. The energy regions instigate the ultraviolet region of the spectrum for Cs2SeCl6, ultraviolet violet edge to the visible region for Cs2SeBr6, and the visible region of Cs2SeI6. Therefore, being a large absorption bandwidth in the visible region of light the Cs2SeI6 is the novel replacement of organic-based perovskites for solar cell applications. The ultraviolet region absorption of Cs2SeCl6 also makes them equally important for sterilizing surgical equipment and other optoelectronic devices.
Absorption coefficients α (ω) also explain the decline of light into the material similar to the imaginary dielectric constant, shown in (c). The critical values of absorption coefficient 3.15, 2.66 and 1.16 eV for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6, respectively. The peak values at 3.6 eV (5.8), 3.15 eV (5.9) and 2.61 eV (6.8) for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6, respectively, show the maximum absorption similar like imaginary dielectric constant. In absorption coefficient, the shifting of peaks towards higher energy region and decreasing of intensities as compared to imaginary dielectric constant is the consequence conversion of dielectric constants into absorption coefficient which depend on frequency. The formula of absorption coefficient contains both real and imaginary parts of dielectric constants.
The refractive index n (ω) elucidates the dispersion of light and transparent behaviour of the material. The pattern of n (ω) and Re ε(ω) are similar and they are related to each other through relation . The calculated values of n (ω) are presented in (d), which also explains the zero-frequency values n (0) and Re ε(0) that satisfied the relation
as shown in . The peak values of refractive index are reported at 3.62 eV (1.6), 3.14 eV (1.61) and 2.56 eV (2.57) for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6 halides, respectively. For the visible region, the refractive index lies between 2 and 3 [Citation32] which confirms Cs2SeI6 is more suitable for visible region operation and its reducing intensity for Cs2SeBr6 and Cs2SeCl6 show the shifting of absorption from the visible region to the ultraviolet region.
The morphology of the materials depends upon the reflection of light which provides information about the roughness of the material surface. The calculated values of reflectivity have been presented in (a). The reflection increases from zero-frequency value and reaches to maximum at 3.62 eV (0.31), 3.11 eV (0.32) and 3.09 eV (0.62) for Cs2SeCl6, Cs2SeBr6 and Cs2SeI6. Therefore, the reflection in the visible region is less as compared to the ultraviolet region. The optical loss of light energy in the form of scattering, heating, etc. has been reported in (b). The optical loss of energy up to 3.0 eV is very negligible. Therefore, the optical characteristic analysis confirms the maximum absorption, less reflectivity and optical loss in the visible region increase the importance of studied materials for solar cells and other optoelectronic applications.
4. Conclusion
In the present article, the electronic, mechanical and optical properties are investigated thoroughly to understand the potential of the studied materials for solar cells and optoelectronics. The value of tolerance factor in the range (0.97–1.0) shows the structural stability while negative formation energy and positive frequencies of phonon dispersion certify thermodynamically stability. The studied materials are also mechanically stable and have ductile nature along with a high melting point. The Cs2SeCl6 has the highest absorption in the ultraviolet region and shifted to the visible region by substituting Cl with Br and I. The variant perovskites Cs2SeI6 is new potential material for visible light solar cells. Additionally, other parameters such as reflection of light and secondly the optical loss possess minimum value in the visible region. Therefore, the highest visible region absorption along with minimum loss of energy makes them interesting compounds for practical applications for solar cells and in optoelectronics.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Funding
References
- Gilani SS, Tariq S, Jamil MI, et al. Elucidating DFT study on structural, electronic, thermal and elastic properties of SrTcO3 by using GGA and mBJ approach. Chin J Phys. 2018;56:308–314.
- Lorenz K, Miranda SMC, Alves E, et al. High pressure annealing of Europium implanted GaN. Mater Devices VII. 2012;8262:82620C.
- Alfaraj N, Mitra S, Wu F, et al. Photoinduced entropy of InGaN/GaN p-i-n double-heterostructure nanowires. Appl Phys Lett. 2017;110:161110.
- Muhammed MM, Alwadai N, Lopatin S, et al. High-efficiency InGaN/GaN quantum well-based vertical light-emitting diodes fabricated on β-Ga2O3 substrate ACS Appl Mater Interfaces. 2017;9:34057.
- Wu F, Sun H, AJia IA, et al. Significant internal quantum efficiency enhancement of GaN/AlGaN multiple quantum wells emitting at ∼350 nm via step quantum well structure design.J Phys D: Appl Phys. 2017;50:245101.
- Tariq S, Mubarak A, Hamioud F, et al. Repercussion of pressure on thermodynamic, optoelectronic, thermoelectric and magneto-elastic rectitude of cubic LaFeO3: quantum DFT perspective. J Alloys Compd. 2020;831:154600.
- Ajia IA, Edwards PR, Pak Y, et al. Generated carrier dynamics in V-Pit-enhanced InGaN/GaN light-emitting diode. ACS Photonics. 2017;5:820.
- Jeong J, Kim M, Seo J, et al. Pseudo-halide anion engineering for α-FAPbI3 perovskite solar cells. Nature. 2021;592:381–385.
- Huang YT, Kavanagh SR, Scanlon DO, et al. Perovskite-inspired materials for photovoltaics and beyond – from design to devices. Nanotechnology. 2021;32:132004.
- Miyasaka T, Kulkarni A, Kim GM, et al. Perovskite solar cells: can we go organic-free, lead-free, and dopant-free? Adv Eng Mat. 2020;10(13):1902500.
- Ajia IA, Edwards PR, Pak Y, et al. Generated carrier dynamics in V-Pit-enhanced InGaN/GaN light-emitting diode. ACS Photonics. 2017;5(3):820–826.
- Xin B, Pak Y, Mitra S, et al. Self-patterned CsPbBr3 nanocrystals for high-performance optoelectronics. ACS Appl Mater Interfaces. 2019;11(5):5223–5231.
- Xin B, Alaal N, Mitra S, et al. Identifying carrier behavior in ultrathin indirect-bandgap CsPbX3 nanocrystal films for use in UV/visible-blind high-energy detectors. Small. 2020;16(43):2004513.
- Saparov B, Sun JP, Meng W, et al. Thin-film deposition and characterization of a Sn-deficient perovskite derivative Cs2SnI6. Chem Mater 2016;28:2315–2322.
- Brik MG, Kityk IV, Phys J. Modeling of lattice constant and their relations with ionic radii and electronegativity of constituting ions of A2XY6 cubic crystals (A = K, Cs, Rb, Tl; X = tetravalent cation, Y = F, Cl, Br, I). Chem Solids. 2011;72:1256–1260.
- Kour R, Arya S, Verma S, et al. Potential substitutes for replacement of lead in perovskite solar cells: a review. Glob Chall. 2019;3(11):1900050.
- Bagnall K, Laidler J, Stewart MAA. Americium chloro-complexes. J Chem Soc A. 1968;1:133–136.
- Morss L, Fuger J. Preparation and crystal structures of dicesium berkelium hexachloride and dicesium sodium berkelium hexachloride. Norg Chem. 1969;8:1433–1439.
- McClure ET, Ball MR, Windl W, et al. Cs2AgBiX6(X = Br, Cl): new visible light absorbing, lead-free halide perovskite semiconductors. Chem Mater. 2016;28:1348–1354.
- Bekenstein Y, Dahl JC, Huang J, et al. The making and breaking of lead-free double perovskite nanocrystals of cesium silver–bismuth halide compositions. Nano Lett. 2018;18:3502–3508.
- Ali MA, Alshahrani T, Murtaza G. Defective perovskites Cs2SeCl6 and Cs2TeCl6 as novel high temperature potential thermoelectric materials. Mater Sci Semicond Process. 2021;127:105728.
- Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev. 1964;136:B864–B871.
- Blaha P, Schwarz K, Madsen GK, et al. WIEN2k: an augmented plane wave + local orbitals program for calculating crystal properties; 2001.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865.
- Tran F, Blaha P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys Rev Lett 2009;102(22):226401–226404.
- Togo A, Tanaka I. First principles phonon calculations in materials science. Scr Mater. 2015;108:1–5.
- Fedorovskiy AE, Drigo NA, Nazeeruddin KM. The role of Goldschmidt's tolerance factor in the formation of A2BX6 double halide perovskites and its optimal range. Small Methods. 2019;1:1900426.
- Sabir B, Murtaza G, Mahmood Q, et al. First principles investigations of electronics, magnetic, and thermoelectric properties of rare earth based PrYO3 (Y = Cr, V) perovskites. Current Appl Phys. 2017;17:1539–1546.
- Karki B, Ackland G, Crain J. Elastic instabilities in crystals from ab initio stress-strain relations. J Phys Condens Matter. 1997;9(41):8579.
- Roknuzzaman M, Ostrikov K, Wang H, et al. Towards lead-free perovskite photovoltaics and optoelectronics by ab-initio simulations. Sci Rep. 2017;7:14025.
- Mahmood Q, Ghrib T, Rached A, et al. Probing of mechanical, optical and thermoelectric characteristics of double perovskites Cs2GeCl/Br6 by DFT method. Mater Sci Semicond Process. 2020;112:105009.
- Cahill DG, Watson SK, Pohl RO. Lower limit to the thermal conductivity of disordered crystals. Phys Rev B. 1992;46:6131.
- Cheng HC, Yu CF, Chen WH. First-principles density functional calculation of mechanical, thermodynamic and electronic properties of CuIn and Cu2In crystals. J Alloys Compd. 2013;546:286–295.
- Anderson OL. A simplified method for calculating the Debye temperature from elastic constants. J Phys Chem Solids. 1963;24:909.
- Hassan M, Arshad I, Mahmood Q. Computational study of electronic, optical and thermoelectric properties of X3PbO (X = Ca, Sr, Ba) anti-perovskites. Semicond Sci Technol. 2017;32:115002.
- Hossain MM, Ali MA, Uddin MM, et al. Origin of high hardness and optoelectronic and thermo-physical properties of boron-rich compounds B6X (X = S, Se): a comprehensive study via DFT approach. J Appl Phys. 2021;129:175109.
- Slack GA. Solid state physics. In Seitz F, Turnbull D, Ehrenreich H, editors. New York: Academic; 1979. Vol. 34, p. 1–71.
- Chen XQ, Niu H, Li D, et al. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallic. 2011;19:1275.
- Ali MA, Hossain MM, Hossain MA, et al. Recently synthesized (Zr1-xTix)2AlC (0 ≤ x ≤ 1) solid solutions: theoretical study of the effects of M mixing on physical properties. J Alloys Compd. 2018;743:146.
- Ali MA, Hossain MM, Jahan N, et al. Newly synthesized Zr2AlC, Zr2 (Al0.58Bi0.42)C, Zr2 (Al0.2Sn0.8)C, and Zr2 (Al0.3Sb0.7)C MAX phases: A DFT based first-principles study. Comput Mater Sci. 2017;131:139.
- Donnell MO, Jaynes ET, Miller JG. Kramers–Kronig relationship between ultrasonic attenuation and phase velocity. J Acoust Soc Am. 1981;69:696.
- Slavney AH, Hu T, Lindenberg AM, et al. A Bismuth-Halide double perovskite with long carrier recombination lifetime for photovoltaic applications. J Am Chem Soc. 2016;138:2138–2141.
- Penn DR. Wave-number-dependent dielectric function of semiconductors. Phys Rev. 1962;128:2093.
- Cai MQ, Yin Z, Zhang MS. First-principles study of optical properties of barium titanate. Appl Phys Lett. 2003;83:2805.