
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
An empirical relationship has been found to evaluate the bulk modulus (B) and shear modulus (G) with the help of two experimental physical quantities (the melting temperature and the crystal ionicity), which are drawn from various pieces of literature. The PVV theory of crystal ionicity and temperature dependence of elasticity are taken into account for the study. The evaluated bulk modulus and shear modulus values are found to be in excellent conformity with experimental and reported values of other researchers. The current research supports the modelling of emerging semiconductor materials and even understanding of their mechanical properties for photovoltaic, spintronics, LiB systems, FLB phenomenon, and superhard material applications.
1. Introduction
A greater part of the natural world and modern technology encircling us is based on solid-state materials. At present, research on the solar cell, high-speed electronics, long-wavelength devices, such as light-emitting diodes, diode lasers, hetero-structure and high electron mobility, electro-optic modulators and photo-detectors, spintronic, Lithium-ion batteries (LiB systems), Fermi level pinning (FLP) phenomenon and superhard materials, has ignited ideas to use zinc-blende compounds as device materials. Most of the binary tetrahedral compounds ANB8-N crystallize either in wurtzite or in zinc-blende structure and have drawn attention [Citation1–10]. In the recent past [Citation11], researchers indulged in the research of zinc-blende structured materials of the crystals type AIIBVI and AIIIBV. This is because of their simplicity in ionic bonding and high symmetry. In these structures, atoms make tetrahedral binding with other elements. These tetrahedral tetrahedrons are present in a cubic form in zinc-blende, despite their hexagonal geometry. In wurtzite, analogous tetrahedral centres are organized in a face-centred cubic (fcc) array, while in the zinc-blende, they are structured in a hexagonal closed-packed (hcp) array [Citation12]. On large scale, during the last quarter of a century, research in physics and chemistry of solids has made great progress in understanding the various properties of these solids [Citation13–22]. The unique omni-triangulated nature of atomic arrangement imparts exclusive physical properties to these materials. The calculations on band structure show that binary compounds are metallic in rocksalt structure, while these are semiconductors in zinc-blende structures [Citation23]. Among the sheer fundamental properties of the material, the research on elastic moduli, such as bulk and shear moduli, is important in understanding the physical structure and mechanical behaviour of materials [Citation24–26]. While research on semiconductors progresses from three dimensions (shape) to two dimensions (thin film) and one dimension (nanowire), the shear modulus becomes more significant. These elastic moduli are interconnected with the help of elastic constants, which are instrumental to almost all properties of the material. Generally, elastic constants are derived from experimental results of the elastic modulus. Steps have been taken over time to understand the elastic, mechanical, electronic, and optical properties of the materials [Citation27,Citation28]. It is well known that diamond and zinc-blende covalent crystals are anisotropic, but the threefold symmetry at the plane [111] in these crystals develops the possibility of isotropic properties on this plane and consequently a way to study isotropic properties of zinc-blende crystals. Nowadays researchers’ attention has turned towards the isotropy of these materials because superhard materials have isotropic lattice and short bond length [Citation29].
In analytical calculations, first-principles calculations are mainly applied to calculate the elastic constants. Because of the complexity and associated cost of first-principles calculations, researchers have always been interested in developing simple empirical relationships for the elastic modulus. Improvements in the modelling of solid-state properties via the density functional theory (DFT) and the easy availability of high-speed computational power have made theoretical theories of these properties somewhat relatively simple from the start. As a result, the determination of solid-state characteristics of binary and ternary compounds has become common, while experimental data are in the spare, but it is difficult to validate published data [Citation30,Citation31]. However, most of the elastic moduli calculations are performed through advanced ab-initio calculations techniques; despite this, one must aware that the justification of the first-principles calculations usually demands subtle familiarity with the inherent feature of the chemical bonding and its characteristics in other materials. However, because of the lengthy methods and also the complexity of computational techniques that use a range of approximations, such a method has always been difficult [Citation32,Citation33]. Many theoretical calculations based on empirical relations have become the preferred method for computational solid-state research in recent years. The availability of state-of-the-art computing facilities made it possible to analyze various physical and structural properties of the materials only through simulation or computation in preference to established experimental methods. The empirical approaches, such as valance, radii, bond length, ionicity, plasmon energy and electronegativity, are often applicable [Citation19,Citation34–38]. This analysis of elastic moduli is a necessary precondition for future investigation of other significant properties. One of the most important requirements for modern materials, whose products are employed in harsh environments, is their ability to withstand high temperatures. This explains why researchers are interested in researching the temperature-dependent variations in these materials’ properties. Recently [Citation39–47], mechanical properties particularly bulk modulus and microhardness of binary tetrahedral compounds have been calculated with the help of melting temperature, crystal ionicity and plasmon energy. Because these semiconductors are sensitive to environmental factors, such as temperature and pressure, they are ideal for artificial devices [Citation6].
Here, we thought it would be of interest to give a relatively straight-forward formula for the bulk and shear moduli of binary tetrahedral semiconductors based only on the concept of ionicity and different melting temperatures of the compounds at ambient temperature (room temperature ∼300 K) and normal pressure (∼0 kbar). Thereafter, the obtained values are consistent with the experimental and reported values by previous researchers. The proposed findings offer important results for zinc-blende structured semiconductors for future experimental work.
2. Theoretical background
Bulk and shear moduli are significant mechanical properties of materials that define material resistance to volume change under compression, in the form of ductility and brittleness of the materials. Recently [Citation48–55], various empirical methods have been mentioned to calculate the bulk modulus of the solids. First, Anderson et al. [Citation48] have put forward an analytical relationship for Bulk modulus in terms of atmospheric pressure and specific volume and applied it to a particular group of compounds. According to Jayaraman et al. [Citation49], the bulk modulus is proportional to the product of ionic charges. Cohen [Citation50] has also suggested the isothermal bulk modulus (at zero pressure) as a function of bond length for rock salt crystals. Based on Phillips and Van Vachten visualization of crystal ionicity [Citation51,Citation52] and philosophical investigations of the bond geometry of zinc-blende materials, Cohen [Citation50] suggested a further relation of the bulk modulus. Lam et al. [Citation53] have also presented an analytical relationship of the bulk modulus with lattice parameters, using local density formalism and the pseudo-potential as a way. Kumar et al. [Citation41] have developed a physical expression for bulk modulus as a function of plasmon energy for binary tetrahedral semiconductors. Al-Douri et al. [Citation4] have suggested two relationships for bulk modulus of IV, III-V, and II-VI semiconductors, one in terms of lattice constant and another relation in terms of transition pressure. Elkenany et al. [Citation7–10] have proposed temperature dependence of bulk modulus for different binary and ternary compounds. Verma et al. [Citation54] have suggested a relationship between bulk modulus as the product of ionic charges and bond length for rock salt and zinc-blende compounds. Verma [Citation55] has also proposed another relationship of bulk modulus as the function of ionic charge product and bond length, and covalency for diamond and zinc-blende structured solids. Kumar et al. [Citation40–42] have used the concept of bond ionicity of Garbato et al. [Citation39] and melting temperature of tetrahedrally coordinated compounds for the determination of bulk modulus [GPa] and found the relation as
(1)
(1) where H [GPa] is the microhardness,
[eV] is the plasmon energy of oscillations,
[K] is the melting temperature and
is the ionicity of the compounds.
It has been obvious that physical parameters are crucial for device development, but none has presented significant results on bulk and shear moduli through different methodologies. These moduli have much melting temperatures and are crystal ionicity-dependent. So, we made a fresh empirical effort to reach accurate results close to experimental values using melting temperature and crystal ionicity of the compound.
Any mechanical change depends upon macro- and micro-variations of the characteristics of the compounds; melting temperature represents this macro-variation and crystal ionicity micro-variation of the compound. As the trend in B, we suggest the following physical expression for the evaluation of Bulk modulus B [GPa] for the binary tetrahedral semiconductors using Phillips’s [Citation56] theory of crystal ionicity based on the quantum mechanical aspect and melting temperature of the compound at ambient temperature and pressure as
(2)
(2) where K1 is a constant; the values of K1 are 0.8 and 0.48 for AIIIBV and AIIBVI compounds, respectively.
Shear modulus defines the stiffness of the material, which measures the resistance of reversible deformation upon the tangential stress. Sahin et al. [Citation57] have shown that shear modulus is related to Poisson ratio and bulk modulus for ternary tetrahedral compounds. Kumar et al. [Citation58] have developed relation of shear modulus depending upon plasmon energy for binary tetrahedral compounds. Kamran et al. [Citation59] have derived a relationship of shear modulus as the function of ionicity and nearest neighbour distance for diamond and zinc-blende structures. Cheng [Citation60] has also proposed various relations on shear modulus in terms of ionicity and bond length for different crystal structures. Verma [Citation55] has shown that shear modulus can be calculated with the help of melting temperature-dependent young’s modulus and atomic volume for diamond-like and zinc-blende structured solids. Varshni [Citation61] has proposed an empirical relation for temperature-dependent elastic moduli of different crystal geometry in varied temperature ranges. Laplanche et al. [Citation62] have suggested an empirical relation in the trend of Varshni’s [Citation61] relation for shear modulus [GPa] for a refractory alloy as
(3)
(3) where
is the elastic modulus at 0 K, and
and tG are constants obtained from fitting experimental data and T [K] temperature over a definite range.
As many researchers proposed that shear modulus depends upon macro- and micro-variations of the characteristics of the compounds in the form of melting temperature and crystal ionicity, these parameters greatly depend on valance electrons and chemical bonds between them. Noting this behaviour, we propose two empirical relations of shear modulus [GPa], considering the bond length for binary tetrahedral semiconductors using Phillips’s [Citation56] theory of crystal ionicity and melting temperature of the compound at ambient temperature and pressure as
(4)
(4)
(5)
(5) where K2 and K3 are constants, and Tm [K] is the melting temperature. The values of K2 = 0.5 and K3 = 0.018 for AIIIBV and AIIBVI compounds, respectively.
3. Results and discussion
The temperature dependence of elastic moduli is significant to improve our understanding of the mechanical behaviour of solids. Elastic moduli usually decline with increasing bond length, as it is present with significant exponents in the equations because ionicity shows distinct patterns with bond length in different crystal systems. Among the major elastic moduli, the bulk modulus explains the stiffness against volume deformation of the material by applied pressure, while volume deformation is directly related to the volume of the constituent atoms. The resistance of constituent atoms to compression is determined by the valence electrons’ gas pressure. The bulk modulus measures the stiffness of the material or the energy necessary to produce deformation. The crystal becomes stiffer as the bulk modulus increases. The shear modulus is a measure of how resistant a material is to reversible defects due to shear stress. The shear modulus also measures the hardness of the material more accurately than the bulk modulus. In the light of the lack of satisfactory and consistent data on the elastic properties of compounds on the one hand and the significance of their knowledge for rigorous examination of the group of physical properties, on the other hand, the aim of the present study is to judiciously calculate and scrutinize associated theoretical and experimental data reported in the literature until now. The bulk and shear moduli of the binary tetrahedral semiconductors have been calculated using the modelled relations (2, 4 and 5). The evaluated values for these compounds are listed in Tables and . The computed results are shown in Figures along with available theoretical and experimental findings. Figure reports model predictions of Equation (2) for melting temperature and ionicity-dependent bulk modulus for the given series of binary compounds and is presented in tables. It is observed that the bulk modulus of the given compounds increases with melting temperature and ionicity.
Figure 1. Plot of bulk modulus against the product of temperature and ionicity. Results obtained are in good accord with exp. and other reported values.
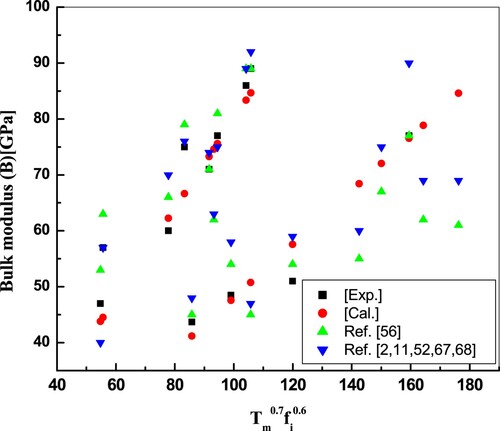
Figure 2. Plot of shear modulus against the product of temperature and ionicity for III-V compounds. Results obtained are in good accord with exp. and other reported values.
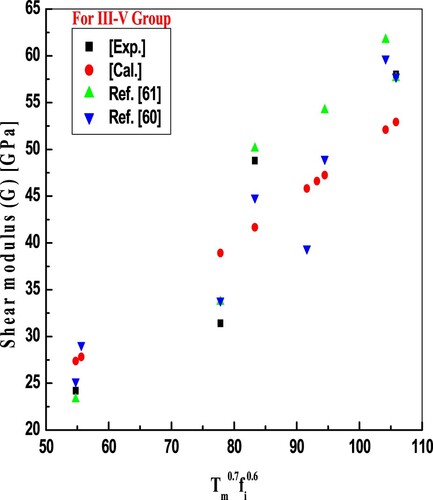
Figure 3. Plot of shear modulus against the product of temperature and ionicity for II-VI compounds. Results obtained are in good accord with exp. and other reported values.
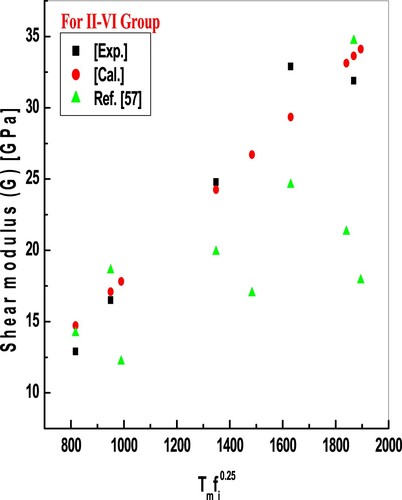
Table 1. Calculated values of B by Equation (2) for binary tetrahedral semiconductors.
Table 2. Calculated values of shear modulus by Equations (4) and (5) for binary tetrahedral semiconductors.
Figures and report model predictions of Equations (3) and (4) for shear modulus and the trend is the same as that of the bulk modulus. From the comparison, we have noticed that evaluated values are in excellent conformity with available experimental and reported values, good accord with experimental values points out that the modelled equations show correlations between melting temperature and elastic moduli. Minimum deviations in calculated values for some compounds from experimental values of elastic moduli in the present endeavour reflect that modelled empirical relations are better than previously proposed relations for the same. Some higher deviations of elastic moduli of these binary compounds are due to larger bond lengths. As the bond length increases heteropolar part of crystal ionicity also decreases and according to the methodology used B and G also reduce, which widens the deviation of results. One more reason for large deviations of moduli values is the lattice mismatch between substrate and deposited films. It is also noted that considered binary compounds are more affected by temperature than ionicity, as observed in calculated values. As, in the present study, we have focused on the melting temperature and crystal ionicity-dependent mechanical properties at ambient temperature and normal pressure, though ionicity depends on energy gaps, which depend upon temperature and pressure [Citation6]. According to Pugh [Citation70], resistance to the plastic deformation is associated with the product of Gb, here constant b is the Burger’s vector, and the product Ba is related to the fracture strength, here constant “a” is related to the lattice parameter. A material of high-value Gb/Ba is brittle. As well, Gb/Ba shows the switching between cohesive and shear strength at the fracture’s crack tip. The constants a and b are specific for a material, the ratio Gb/Ba can be reduced to G/B. The G/B ratio can be used to determine the fragility and brittle-ductile transition characteristics of the materials using the developed formulae for B and G, which also show the ionic and covalent character of the materials [Citation58,Citation71]. Since the ionicity is so significant in the covalent material’s brittle behaviour, comparatively high ionicity results in a G/B ratio of greater than or equal to 0.5, which is exclusive to brittle materials [Citation65,Citation72]. The major improvement in the present theory is the uncomplicated relation, which can correlate melting temperature and ionicity values and predict the values of B and G of unknown semiconductors in analogous series.
4. Summary and conclusions
Till now, various models of finding elastic moduli of semiconductors have been evolved by theoretical researchers, but small variations in unit cell geometry had always been weighed over the correctness of determining these moduli. Therefore, we have developed empirical formulas of bulk and shear moduli of covalent binary compounds and obtained their results, which are in good agreement with the previously published theoretical and experimental data. This study helps in the modelling of emerging semiconductor materials of the studied groups and finding their mechanical properties for device applications. This model can also easily be extended to other more complex crystals, of which work is in progress.
Acknowledgements
The authors are thankful to the referee for the valuable comments that have been useful in revising the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Shay JL, Wernick JH. Ternary chalcopyrite semi-conductors: growth, electronic properties and applications. Oxford: Pergamon Press; 1975.
- Tutuncu HM, Bagci S, Srivastava GP, et al. Structural and dynamical properties of zinc-blende GaN, AlN, BN, and their (110) surfaces. Phys Rev B. 2005;71:195309.
- Marad AE, Kanoun MB, Merad G, et al. Full-potential investigation of the electronic and optical properties of stressed CdTe and ZnTe. Mater Chem Phys. 2005;92:333.
- Al-Douri Y, Abid H, Aourag H. Correlation between the bulk modulus and the transition pressure in semiconductors. Mater Lett. 2005;59:2032.
- Degheidy AR, Elkenany EB. Structural and electronic properties of Si1−xGex binary semiconducting alloys under the effect of temperature and pressure. Semiconductors. 2013;47:1283.
- Degheidy AR, Elkenany EB. Temperature and pressure dependence of the electronic and optical properties of GaxIn1−xAsyP1−y matching different substrates. Phys B. 2015;456:213.
- Degheidy AR, Elkenany EB, Alfrnwani OA. Temperature and pressure dependence of elastic constants and related parameters for InP semiconductor. Silicon. 2017;9:183.
- Degheidy AR, Elkenany EB, Alfrnwani OA. Mechanical properties of AlPxSb1-x semiconductor alloys under the effect of temperature and pressure. Comp Cond Mat. 2018;16:e00310.
- Degheidy AR, Elkenany EB, Madkour M, et al. Temperature dependence of phonons and related crystal properties in InAs, InP and InSb zinc-blende binary compounds. Comp Cond Mat. 2018;16:e00308.
- Elkenany EB. Energy band structure, acoustic velocities, optical phonon frequencies and mechanical properties of InP1-xSbx alloys under temperature and pressure. Infr Phys Tech. 2021;115:103720.
- Mujica A, Rubio A, Munoz A, et al. High-pressure phases of group-IV, III–V, and II–VI compounds. Rev Mod Phys. 2003;75:863.
- Jain M. Diluted magnetic semiconductors. Singapore: Word Scientific; 1991.
- Xia Y, Hegde VI, Pal K, et al. High-throughput study of lattice thermal conductivity in binary rocksalt and zinc blende compounds including higher-order anharmonicity. Phy Rev X. 2020;10:041029.
- Boubendira K, Bendaif S, Nemiri O, et al. Fundamental properties of zinc-blende AlSb, BSb and their Al1-xBxSb ternary alloys. Chin J Phys. 2017;55:1092.
- Pal S, Tiwari RK, Gupta DC, et al. Inter atomic force constants of binary and ternary tetrahedral semiconductors. Semiconductors. 2016;50:795.
- Tripathy SK, Patnaik A. Optical and electronic properties of some semiconductors from energy gaps. Opt Mat. 2016;53:123.
- Li X, Wang H, Fu Z. A simple bulk modulus model for crystal materials based on the bond valence model. Phys Chem Chem Phys. 2017;19:22177.
- Lv P, Yang S, Liu C, et al. Pressure-induced emission enhancements and ripening of zinc blende cadmium selenide nanocrystals. J Phys Chem C. 2019;123:15339.
- Huang C, Feng J, Zhou J, et al. Ultra-high-temperature ferromagnetism in intrinsic tetrahedral semiconductors. J Am Chem Soc. 2019;141:12413.
- Tung RT, Kronik L. Fermi level pinning for zinc-blende semiconductors explained with interface bonds. Phys Rev B. 2021;103:085301.
- Gupta RC, Singh K, Verma AS. Optoelectronic properties of ternary tetrahedral semiconductors. East Eur J Phys. 2021;1:89.
- Feng X, Xiao J, Wen B, et al. Temperature-dependent hardness of zinc-blende structured covalent materials. Sc Chn Mater. 2021;64:2280.
- Salami L, Meradji H, Ghemid S, et al. Phase stability, pressure-induced phase transition and electronic properties of AlX (X = P, As and Sb) compounds from first principle calculations. Phase Transitions. 2020;93:843.
- Pawar P, Shukoor V A, Singh S. Pressure induced phase transition of AlX (X = P, As) compounds under effect of temperature. Mater Today Proc. 2018;5:2474.
- Bajpai G, Srivastava T, Nasir M, et al. A comprehensive theoretical and experimental study on structural and mechanical properties of Si doped ZnO. Scr Mater. 2017;135:1.
- Ha VTT, Hung VV, Hanh PTM, et al. Structural and thermomechanical properties of the zinc-blende AlX (X = P, As, Sb) compounds. Int J Mod Phys B. 2017;31:1750141.
- Daoud S, Bencheikh A, Belagraa L. Quasi-linear correlation between high-frequency and static dielectric constants in II-VI and III-V semiconductors. Int J Phy Res. 2016;5(1):4.
- Kumar V, Singh BP, Chandra S. Optical, elastic and thermal properties of ZB-AlN semiconductor from first-principle calculations. Ind J Phys. 2017;91(4):445.
- Ravindran NM, Chen D. Semiconductors: other miscellaneous semiconductors and related binary, ternary, and quaternary compounds, Vol. 481. Springer; 2019.
- Wang SQ, Ye HQ. Ab initio investigation of the pressure dependences of phonon and dielectric properties for III–V semiconductors. J Phys Condensed Matter. 2005;17:4475.
- Derby B. Correlations for single-crystal elastic constants of compound semiconductors and their representation in isomechanical groups. Phys Rev B. 2007;76:054126.
- Verma AS. Bond-stretching force constant of AIBIIIC2V I and AIIBIVC2V chalcopyrite semiconductors. Solid State Comm. 2009;149:1236.
- Kumar V, Shrivastava AK, Banerji R, et al. Debye temperature and melting point of ternary chalcopyrite semiconductors. Solid State Comm. 2009;149:1008.
- Kumar V, Sastry BSR. Heat of formation of ternary chalcopyrite semiconductors. J Phys Chem Solids. 2005;66:99.
- Pauling L. The nature of chemical bond. 3rd ed. Ithaca (NY): Cornell University Press; 1960.
- Daoud S. Empirical study of elastic properties of BX (X = As, Sb) materials. Int J Sci World. 2014;3(1):37.
- Gupta RC, Verma AS, Singh K. Empirical relation for electronic and optical properties of binary tetrahedral semiconductors. East Eur J Phys. 2021;1:89.
- Wang J, Chen H, Wei SH, et al. Materials design of solar cell absorbers beyond perovskites and conventional semiconductors via combining tetrahedral and octahedral coordination. Adv Mater. 2019;31:1806593.
- Garbato L, Rucci A. Phil Mag. 1977;35:1681, 1985.
- Prasad GM. Microhardness of AN−1 BN + 1 C28−N chalcopyrite semiconductors. Physica Stat Solidi. 1992;170:77.
- Kumar V, Prasad GM, Chetal AR, et al. Microhardness and bulk modulus of binary tetrahedral semiconductors. J Phys Chem Solids. 1996;57:503.
- Kumar V, Prasad GM, Chandra D. Microhardness of AN−1 BN + 1 C28−N Chalcopyrite Semiconductors. Ind J Pure App Phys. 1994;32:272.
- Zakarian D, Khachatrian A, Firstov S. Universal temperature dependence of Young’s modulus. Met Powder Rep. 2019;74:204.
- Daoud S. Electronic polarizability of ABC2 chalcopyrite semiconductors. Semi Phys. 2019;22:404.
- Kurchin RC, Gorai P, Buonassisi T, et al. Structural and chemical features giving rise to defect tolerance of binary semiconductors. Chem Mater. 2018;30:5583.
- Algarni H, Al-Hagan OA, Bouarissa N, et al. Pseudopotential calculations of AlSb under pressure. Spectro Acta Part A. 2018;190:215.
- Yin Y, Chen G, Ye H, et al. Structural and chemical features giving rise to defect tolerance of binary semiconductors. Let J Exp Fron Phys. 2016;114:36001.
- Anderson OL, Nafe JE. The bulk modulus-volume relationship for oxide compounds and related geophysical problems. Geophys J Res. 1965;70:3951.
- Jayaraman A, Batlogg B, Maines RG, et al. Effective ionic charge and bulk modulus scaling in rocksalt-structured rare-earth compounds. Phys Rev B. 1982;26:3347.
- Cohen ML. Calculation of bulk moduli of diamond and zinc-blende solids. Phys Rev B. 1985;32:7988.
- Phillips JC, Van Vechten JA. Spectroscopic analysis of cohesive energies and heats of formation of tetrahedrally coordinated semiconductors. Phys Rev B. 1970;2:2147.
- Phillips JC, Van Vechten JA. Nonlinear optical susceptibilities of covalent crystals. Phys Rev B. 1969;183:709.
- Lam PK, Cohen ML, Martinez G. Analytic relation between bulk moduli and lattice constants. Phys Rev B. 1987;35:9190.
- Verma AS, Bhardwaj SR, Phys J. Correlation between ionic charge and ground-state properties in rocksalt and zinc blende structured solids. Cond Matt. 2006;18:8603.
- Verma AS. Elastic moduli and brittleness of diamondlike and zinc blende structured solids. Mat Chem Phys. 2012;135:106.
- Phillips JC. Ionicity of the chemical bond in crystals. Rev Mod Phys. 1970;42:317.
- Sahin S, Ciftci YO, Colakoglu K, et al. First principles studies of elastic, electronic and optical properties of chalcopyrite semiconductor ZnSnP2. J Alloys Compd. 2012;529:1.
- Kumar V, Singh JK, Prasad GM. Elastic properties of elemental, binary and ternary semiconductor materials. Ind J Pure Appl Phys. 2015;53:429.
- Kamran S, Chen K, Chen L. Semiempirical formulae for elastic moduli and brittleness of diamondlike and zinc-blende covalent crystals. Phys Rev B. 2008;77:094109.
- Cheng D. Mechanical, electronic and optical properties of multi-ternary semiconductor alloys [Ph.D. Thesis]. Newark: The State University of New Jersey; 2013.
- Varshni YP. Temperature dependence of the elastic constants. Phys Rev B. 1970;2:3952.
- Laplanche G, Gadaud P, Perriere L, et al. Temperature dependence of elastic moduli in a refractory HfNbTaTiZr high-entropy alloy. J Alloys Comp. 2019;799:538.
- Kumar V, Sastry BSR. Thermal expansion coefficient of binary semiconductors. Cryst Res Tech. 2001;36:565.
- Omar MS. Lattice thermal expansion for normal tetrahedral compound semiconductors. Mater Res Bull. 2007;42:319.
- Pettifor DG. Theoretical predictions of structure and related properties of intermetallics. Mater Sci Technol. 1992;8:345.
- Wang SQ, Ye HQ. Plane-wave pseudopotential study on mechanical and electronic properties for IV and III-V crystalline phases with zinc-blende structure. Phys Rev B. 2002;66:235111.
- Kim K, Lambrecht WRL, Segall B. Elastic constants and related properties of tetrahedrally bonded BN, AlN, GaN, and InN. Phys Rev B. 1996;53:16310.
- Kumar V, Shrivastava AK, Jha V. Bulk modulus and microhardness of tetrahedral semiconductors. J Phys Chem Solids. 2010;71:1513.
- Al-Douri Y, Abid H, Aourang H. Correlation between the bulk modulus and the charge density in semiconductors. Physica B. 2001;305:186.
- Pugh SF. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos Mag. 1954;45:823.
- Greaves GN, Greer AL, Lakes RS, et al. Poisson's ratio and modern materials. Mater. 2011;10:823.
- Pettifor DG, Cottrell AH. Electron theory in alloy design. London: The Institute of Material; 1992.