
Abstract
Various 2-substituted-1,3-dithiane oxides (1-oxide and 1,3-dioxide) have been metalated and reacted for the first time with a trialkylborane (trioctylborane). The 2-chloro-1,3-dioxide results in migration of an octyl group from boron to carbon with the displacement of chloride and gives nonanoic acid after oxidation, but there is no evidence for a second migration involving displacement of a sulfenate group. The reaction involving lithiation of the 2-methoxy-1-oxide results in two migrations, with the displacement of both the methoxy group and the thiolate unit of the dithiane ring, giving dioctyl ketone after oxidation, but the yield is low, primarily because thiophilic addition of the lithiating agent predominates over lithiation. Again, there is no evidence for the displacement of the sulfenate unit. However, the intermediate prior to oxidation can be treated with trifluoroacetic anhydride to induce a Pummerer rearrangement, and the presumed trifluoroacetoxyalkylthiolate group then acts as a novel leaving group and is displaced, resulting in trioctylmethanol on oxidation, but the yield is again very low.
GRAPHICAL ABSTRACT
1. Introduction
Organoboranes are common reagents that enable many useful synthetic transformations [Citation1–4]. Many of those reactions involve the 1,2-migration of an alkyl group from boron to an electrophilic center bearing a leaving group at the α-position. Although alkylboron compounds are quite stable as a result of the low polarity of the C–B bond, they can be oxidized easily [Citation5,Citation6]. Oxidation following a 1,2-migration reaction leads to the production of various products, including carbonyl compounds and alcohols [Citation7]. Various factors affect the migration of groups in such reactions, including the steric bulk of the groups around the boron atom, which can play an important role in controlling migration [Citation8].
Reactions of trialkylboranes with various trisubstituted methanes such as chloroform (CHCl3), dichlorofluoromethane (CHCl2F), chlorodifluoromethane (CHClF2) and 1,1-dichloromethyl methyl ether (DCME) in the presence of a strong base result in the transfer of all three alkyl groups from boron-to-carbon in a single process [Citation9,Citation10]. Even trialkylboranes having a tertiary alkyl group, such as a tert-butyl or thexyl moiety, on reaction with DCME and lithium triethylcarboxide at 25°C, transfer all three groups successfully [Citation9,Citation10]. A reagent with three different leaving groups attached to a central carbon atom could, in principle, be used as an alternative to DCME, opening up possibilities for asymmetric induction to generate enantiomerically enriched chiral tertiary alcohols. Compounds having two sulfur-containing leaving groups have been used successfully to perform up to two 1,2-boron to carbon migrations [Citation11–15], and in principle, a third leaving group could be incorporated to allow a third migration. A potential advantage of using sulfur-based leaving groups might be that stereoselectivity could be controlled, as it can, for example, in reactions of various electrophiles with metalated 1,3-dithiane oxides [Citation16–21]. Such reagents might be able to offer possibilities for the generation of appropriately substituted chiral reagents for reactions with trialkylboranes. However, some basic studies are needed in order to underpin such possibilities.
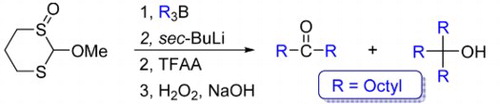
We have been extensively involved in the development of useful synthetic methods that utilize boron and lithium intermediates [Citation22–31]. We therefore turned our attention towards reactions between trialkylboranes and appropriate lithiated 1,3-dithiane oxides derived from 1, including trans-1,3-dithiane-1,3-dioxide 2, a mixture of cis- and trans-2-chloro-1,3-dithiane-1,3-dioxides 3, and 2-substituted 1,3-dithiane-1-oxides 4–6 (Figure ). Our intention was to investigate what kinds of reactions, if any, would take place. We now report the results.
Figure 1. 1,3-Dithiane oxide derivatives 1–6.
2. Results and discussion
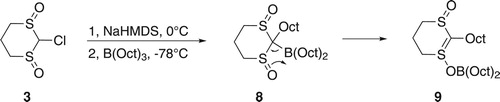
Reaction of trans-1,3-dithiane-1,3-dioxide 2 (Figure ) and n-butyllithium (n-BuLi) in a mixture of pyridine and tetrahydrofuran (THF), followed by the addition of an aldehyde as an electrophile, gives the corresponding alcohol as a mixture of two diastereoisomers in a ratio of around 1:2 [Citation32]. The reaction of 2 and n-BuLi (two mole equivalents), followed by the addition of tri-n-octylborane at –78°C and then HgCl2 was attempted. After work-up, 1-octanol was the only product, indicating that no migration had taken place under the conditions tried. Possibly, no boron–carbon adduct was produced as a result of the steric hindrance caused by the two sulfoxide groups. Alternatively, the sulfoxide group might not be a good leaving group. In order to try to distinguish these possibilities, the reaction of trioctylborane and metalated 3, which contains a good leaving group (Cl), was attempted.
Metalation of 3 (Figure ) was attempted using sodium bis(trimethylsilyl)amide (NaHMDS) at 0°C. The metalated intermediate was allowed to react with trioctylborane at −78°C followed by oxidation using a basic solution of hydrogen peroxide (H2O2; Scheme 1). Following work-up, nonanoic acid (7) was obtained in 50% yield along with 1-octanol. The GC–MS spectrum showed the presence of traces of dioctyl ketone as a result of two migrations, but the amount was not significant. Attempts to encourage the second migration by the use of a higher temperature were not successful.
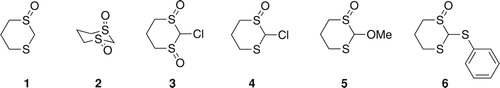
Figure 2. Structures of 12 and 13.
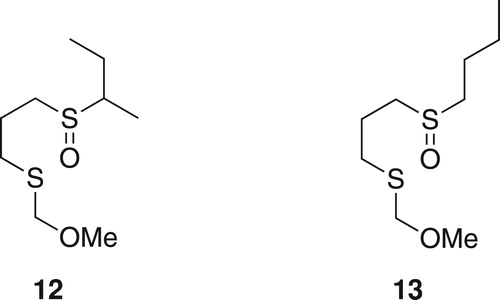
Scheme 1. Reaction of 3 and trioctylborane to produce nonanoic acid 7.
Clearly, an octyl group had replaced the chlorine, although apparently in only moderate yield. It is possible that steric hindrance caused by the two sulfoxide groups inhibited the complete formation of the B–C adduct. The failure to give more than one boron to carbon migration could suggest that the sulfoxide group might not act as a good leaving group so that the reaction stops after the first migration. However, it could also be that the intermediate 8 formed after the first migration rearranges to a more stable boron enolate-like compound 9 (Scheme 2), in a manner similar to that which occurs in reactions of trialkylboranes with anions of α-bromocarbonyl compounds [Citation33,Citation34], as we have experienced previously in a phenylsulfoxide system [Citation24]. The product of the rearrangement 9 would no longer be of the type that is susceptible to B–C migrations.
Scheme 2. Possible rearrangement of the intermediate after the first migration step.
Some insight into such possibilities might be provided by reactions of 2-substituted-1,3-dithiane-1-oxides 4–6 (Figure ) because after the first migration, there would still be another potential leaving group (a thiolate anion), even if the sulfoxide moiety is not a good enough leaving group. First, we decided to attempt metalation of 4 followed by reaction with several electrophiles (e.g. iodoethane, 3,4-dimethoxybenzaldehyde and benzaldehyde) to ensure that appropriate conditions for metalation had been found. Several bases (e.g. n-BuLi, NaHMDS and LDA) at different temperatures (0°C, −78°C and −100°C) were used. However, no new products were identified under the conditions used. One explanation for such observations is that the metalated intermediate of 4 is not stable under the conditions attempted. Therefore, in order to stabilize the anion produced in-situ, the chlorine in 4 was replaced by a methoxy group (i.e. 5). Compound 5 (Figure ) was a mixture of two diastereomers in a ratio of 81:19.
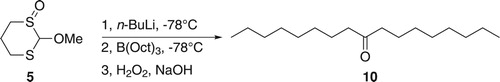
The reaction of 5 with n-BuLi followed by trioctylborane (Scheme 3) was investigated to test for possible boron-to-carbon 1,2-migrations. Indeed, the use of 5 led to the formation of dioctyl ketone (10; Scheme 3), which was isolated in 13% yield along with 1-octanol (72% of all octyl groups of tri–n–octylborane). However, no product of a triple migration was seen.
Scheme 3. Reaction of 5 and trioctylborane to produce 9-heptadecanone (dioctyl ketone), 10.
Although it was not clear why the yield of 10 was so low, it was encouraging that some dioctyl ketone had been formed, indicating that not only the methoxy group but also a second group, presumably the thiolate group, could be displaced by an octyl group during the process. However, it was disappointing that a third migration, presumably involving displacement of a sulfoxide moiety, did not take place. Whether this was due to the inability of the sulfoxide to act as a leaving group or whether the intermediate formed after two migrations could undergo a rearrangement akin to that shown in Scheme 2 to prevent a further migration was unclear. Therefore, before attempting to improve the yield of 10, it was important to understand whether the intermediate after two migrations was still capable of being induced to undergo a further migration.
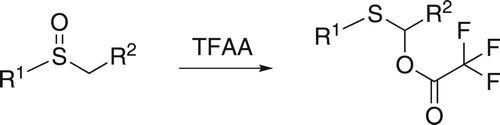
The Pummerer rearrangement is a common way to convert an alkyl sulfoxide into an α-acyloxyalkyl sulfide by the use of acetic anhydride or trifluoroacetic anhydride (TFAA; Scheme 4) [Citation35].
Scheme 4. Pummerer rearrangement induced by TFAA.
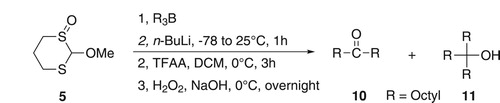
An α-trifluoroacetoxyalkylthiolate group should be a better leaving group than a simple alkylthiolate group. Therefore, if the second octyl migration had involved the displacement of the thiolate group and the intermediate thus produced had not undergone a rearrangement akin to that shown in Scheme 2, a Pummerer rearrangement conducted on the intermediate might induce the third migration. In order to test this possibility, the reaction of 5 with n-BuLi and trioctylborane was conducted as before, but then excess TFAA (1.3 mole equivalents) was added at 0°C and the mixture was stirred for 3 h. Following oxidation of the mixture and work-up, a mixture of 10 (4%), trioctylmethanol (11) (6%), and 1-octanol (69% of all octyl groups of tri-n-octylborane) was produced (Scheme 5). A possible mechanism for the formation of 11 is shown in Scheme 6.
Scheme 5. Induction of the third migration via a Pummerer rearrangement.
Scheme 6. A possible mechanism for the formation of 11.
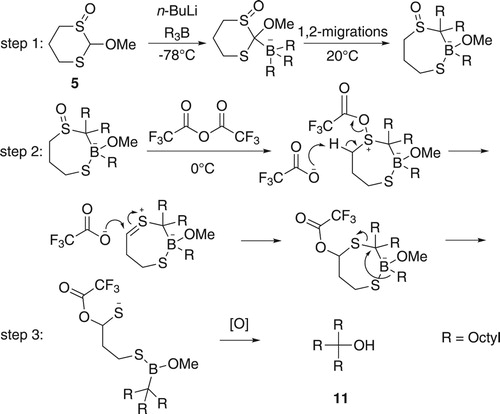
The combined yields of 10 and 11 were not very different from the yield of 10 achieved when no Pummerer rearrangement was incorporated. Not, all of the double migrated product was converted into a triple migrated product, which could indicate that the Pummerer rearrangement had not gone to completion under the conditions attempted or that some of the double migration intermediates had undergone a rearrangement akin to that in Scheme 2. However, the most significant result was that triple migration could be induced, and before investigating whether the conversion of double migrated product into the triple migrated product could be improved, it was important to try to improve the yield of the double migration product itself.
It was thought that the low yield of 10 using n-BuLi as the lithiating agent might be due to the poor formation of 2-lithio-2-methoxy-1,3-dithiane-1-oxide with this reagent. Therefore, other lithium reagents were used in attempts to improve the yield of 10. The GC yields of 10 obtained under the conditions used are reported in Table .
Table 1. The GC yield of 10 according to Scheme 3 but using different lithium reagents.
The use of 1.1 mole equivalents of sec-BuLi led to the production of 10 in 28% yield (Table ; Entry 4) compared to 16% when n-BuLi (1.1 mole equivalents) was used (Table ; Entry 1). To our surprise, tert-BuLi, which is a stronger base compared with both n-BuLi and sec-BuLi, led to the formation of 10 in only 12% yield (Table ; Entry 2). sec-BuLi is less sterically hindered than tert-BuLi and is a stronger base than n-BuLi. Since sec-BuLi provided the highest yield, it was of interest to test the effect of the quantity of sec-BuLi on the yield of 10. Therefore, several experiments were carried out using different mole equivalents (1.0–1.8) and the GC yields of 10 are recorded in Table (Entries 3–7), which showed that the yield of 10 peaked at 31% when sec-BuLi (1.2 mole equivalents) was used (Table ; Entry 5). Several other attempts were made to improve the yield of 10 with sec-BuLi. For example, cooling the reaction mixture to −100°C led to a similar yield to that obtained at −78°C (31%).
It was suspected that the lithium intermediate of 5 was not produced cleanly under the conditions attempted. To test this theory, lithiation of 5 was attempted using sec-BuLi (1.2 mole equivalents) at −78°C followed by protonation using aqueous ammonium chloride (NH4Cl), which would regenerate 5 in quantitative yield if there were to be clean formation of the lithiated derivative. The crude product was purified, and a new product was identified as 12 (Figure ; See Experimental Section for characterization). Compound 12 would have resulted from the initial thiophilic addition of sec-BuLi to the sulfoxide group followed by ring-opening, and it was isolated in 49% yield as a 1:1 mixture of two diastereoisomers. Such a result explains why the yield of 10 dropped to 0% when a large excess of sec-BuLi (1.8 mole equivalents) was used.
Reaction of 5 with n-BuLi followed by protonation led in the same way to the production of 13 (Figure ) in 40% yield. On the other hand, the reaction of 5 with tert-BuLi under similar reaction conditions did not lead to any new products and the starting material was recovered. In the latter case, both thiophilic addition and deprotonation might be restricted due to the hindrance of the tert-butyl group.
Next, our attention turned to the use of less nucleophilic lithium reagents such as lithium diisopropylamide (LDA), lithium tetramethylpiperidide (LiTMP) and lithium bis(trimethylsilyl)amide (LiHDMS). First, we attempted reaction of 5 and LDA followed by protonation. Only starting material was recovered, indicating that 5 did not undergo thiophilic addition such as was seen with sec- and n-BuLi. However, when the lithiated intermediates were treated with trioctylborane the yields of 10 were still disappointingly low (Table ; Entries 8–12). The best yield of 10 with the lithium amide reagents was 30% when the reaction was carried out with LDA (1.1 equivalents) at 0°C (Table ; Entry 9). The use of a superbase (Schlosser’s reagent, LICKOR [Citation36]) provided no improvement.
Finally, we investigated the use of reagent 6, in the hope that the sulfur atom of the SPh group would provide better stabilization of the lithiated species than the OMe group and might lead to a greater preference for lithiation over thiophilic addition, leading to an improvement in the yield of 10. However, on reaction of 6 with trioctylborane, 10 was obtained in only 4% yield along with 1-octanol as the main product. Therefore, no further studies were conducted.
3. Conclusion
Reactions of lithiated dithiane oxides, generated using different bases, with trialkylboranes, followed by oxidation, have been investigated. The use of 2-chloro-1,3-dithiane-1,3-dioxide led to only one migration, producing nonanoic acid in moderate yield (50%). 2-Methoxy-1,3-dithiane-1-oxide led to two migrations, giving dioctyl ketone in up to 31% yield and a third migration could be induced, leading eventually to trioctylmethanol, by addition of trifluoroacetic anhydride to bring about a Pummerer rearrangement prior to oxidation. Various attempts have been made to try to find conditions under which the yields could be improved, but no conditions were found that would render the reactions useful for synthetic methodology. The major problem appears to be in bringing about quantitative formation of an appropriate lithiated sulfoxide species, but if this issue can be resolved, the reactions would potentially offer an approach for the synthesis of chiral tertiary alcohols.
4. Experimental section
4.1. General
Chemicals, reagents and solvents were purchased from Aldrich Chemical Company. The solvents were purified using standard procedures [Citation37]. The concentration of BuLi was determined prior to use [Citation38]. LDA-LICKOR superbase was prepared based on a literature procedure [Citation36]. Bruker AV400 or AV500 spectrometers were used to record the 1H (400 or 500 MHz) and 13C NMR (100 or 125 MHz) spectra. Low- and high-resolution mass spectra were recorded on a Waters GCT Premier spectrometer and a Waters LCT Premier XE instrument, respectively. GC measurements were carried out using a Shimadzu GC-2014 gas chromatograph fitted with a ZB-5 column (30, 0.32 mm inner diameter, 1.0 μm film thickness). The carrier gas was He at 69.3 kPa, and a split injection mode was used. The oven temperature was increased from 70°C to 260°C at 6°C min−1 and then held for 4 min. Tetradecane was used as an external standard to allow quantification of products.
4.2. Preparation of 1,3-dithiane-1-oxide (1) [39]
Sodium periodate (1.07 g, 5 mmol) in H2O (35 mL) was added slowly over 30 min to a stirred solution of 1,3-dithiane (0.60 g, 5.0 mmol) in MeOH (40 mL) in a round-bottom flask (250 mL) at a temperature less than 20°C. The mixture was stirred for 30 min, and the solid formed was collected by filtration, washed with dichloromethane (DCM; 3 × 20 mL) and the filtrate and washings were combined and concentrated under reduced pressure. The residue obtained was extracted with DCM (3 × 20 mL), the extract was dried (MgSO4) and filtered, and the solvent was removed under reduced pressure to give 1 (0.58 g, 85%) as a colorless solid, m.p. 85–86°C (lit [Citation39]. 86–87°C). 1H NMR (400 MHz; CDCl3) δ 3.99 (d, J = 12.7 Hz, 1H), 3.63 (d, J = 12.7 Hz, 1H), 3.39–3.24 (m, 1H), 2.71–2.43 (m, 4H), 2.32–2.10 (m, 1H). 13C NMR (125 MHz; CDCl3) δ 52.9, 50.5, 28.3, 27.1.
4.3. Preparation of trans-1,3-dithiane-1,3-dioxide (2) [40]
Sodium periodate (5.35 g, 25 mmol) was added to a suspension of 1,3-dithiane (1.20 g, 10 mmol) in a mixture of MeOH (35 mL) and H2O (3.5 mL). The mixture was stirred for 96 h then Me2S (0.75 mL, 10 mmol) was added, and the mixture stirred for 30 min. The solvent was removed under reduced pressure, and the white solid obtained was extracted with a mixture of acetone and EtOH (5:1 by volume). The extract was passed through a short pad of silica with a mixture of acetone and EtOH (5:1 by volume) as the eluent. The solvent was removed under reduced pressure, and the cis and trans mixture was purified by flash column chromatography (silica; acetone) to give 2 (0.94 g, 62%) as a colorless solid, m.p. 171–172°C (lit [Citation40]. 170–171°C). 1H NMR (500 MHz; DMSO-d6) δ 4.34 (s, 2H), 3.27–3.15 (m, 2H), 3.02–2.91 (m, 2H), 2.66–2.15 (m, 2H). 13C NMR (125 MHz; DMSO-d6) δ 61.8, 47.6, 14.9.
4.4. Preparation of 2-chloro-1,3-dithiane-1,3-dioxide (3) [21]
N-Chlorosuccinimide (147 mg, 1.1 mmol) was added to a stirred solution of 2 (152 mg, 1.0 mmol) in dry DCM (10 mL). The mixture was stirred at 20°C for 23 h and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (silica; EtOH:EtOAc = 1:9 by volume) to afford 3 (158 mg, 85%) as a colorless solid, m.p. 139–141°C (lit [Citation21]. 141–142°C). 1H NMR (500 MHz; CDCl3) δ 5.93 (s, 1H), 3.46–3.08 (m, 3H), 2.97 (m, 1H), 2.85–2.60 (m, 1H), 2.48–2.16 (m, 1H). 13C NMR (125 MHz; CDCl3) δ 75.3, 45.4, 41.5, 14.5.
4.5. Synthesis of 2-chloro-1,3-dithiane-1-oxide (4)
N-Chlorosuccinimide (147 mg, 1.1 mmol) was added to a stirred solution of 1 (136 mg, 1.0 mmol), prepared as described in Section 4.2, in dry DCM (10 mL). The mixture was stirred at 20°C for 23 h and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (silica; EtOAc:Et2O = 1:10 by volume) to give 4 (0.103 g, 60%) as a mixture of two diastereomers (58:42 ratio) as a light yellow solid, m.p. 48–70°C. νmax (NaCl film) 2995, 2940, 2844, 1423 cm–1. 1H NMR (400 MHz; CDCl3) the signals for the two isomers overlapped except for the CHCl proton; δ 5.90 (s, 1H of major isomer), 5.49 (s, 1H of minor isomer), 3.33–3.04 (m, 3H), 3.04–2.89 (m, 2H), 2.87–2.74 (m, 1H), 2.68–2.55 (m, 1H), 2.46–2.17 (m, 4H), 1.83–1.64 (m, 1H). 13C NMR (100 MHz; CDCl3) major isomer: δ 74.0, 45.9, 28.9, 23.2; minor isomer: 70.2, 41.0, 29.7, 23.4. MS (EI) m/z (%) 172 (37ClM+, 12%), 170 (35ClM+, 36), 135 (16), 106 (100), 90 (95), 64 (30). HRMS: Found for 35ClM+ (C4H7ClOS2): 169.9630, calculated: 169.9627.
4.6. Synthesis of 2-methoxy-1,3-dithiane-1-oxide (5)
MeOH (10 mL) was added to sodium metal (14 mg, 0.6 mmol) under nitrogen in an ice bath and stirred for 10 min. The solution obtained was transferred in a dropwise manner through a syringe to a stirred cooled (0°C) solution of 4 (103 mg, 0.6 mmol) in dry THF (4 mL). The mixture was allowed to warm to room temperature and stirred for 1 h. The solvents were removed under reduced pressure, and the solid obtained was extracted with CHCl3 (3 × 10 mL). The combined extracts were washed with brine, separated and dried (MgSO4). The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography (silica; EtOAc:Et2O = 1:10 by volume) to yield 5 (70 mg, 70%), as a mixture of two diastereomers in a ratio of 81:19, as light yellow oil. νmax (neat) 2935, 2907, 2831, 1424, 1084 and 1029 cm–1. 1H NMR (400 MHz; CDCl3) δ 5.32 (s, 1H of major isomer), 5.01 (s, 1H of minor isomer) 3.75 (s, 3H of major isomer), 3.64 (s, 3H of minor isomer), 3.36 (td, J = 12.8, 2.8 Hz, 1H of major isomer), 3.12 (t, J = 6.6 Hz, 1H of minor isomer), 3.01–2.95 (m, 1H of each isomer), 2.87–2.80 (m, 1H of each isomer) 2.69–2.54 (m, 1H of minor isomer), 2.42–2.20 (m, 3H of major isomer and 2H of minor isomer). 13C NMR (100 MHz; CDCl3) major isomer: δ 90.2, 59.5, 45.2, 29.3, 22.2; minor isomer: δ 93.2, 58.7, 42.2, 31.1, 23.2. MS (EI) m/z (%) 166 (M+, 68%), 135 (5), 106 (100), 90 (98) and 64 (95). HRMS: Found for M+ (C5H10O2S2): 166.0125, calculated: 166. 0122.
4.7. Synthesis of 2-thiophenyl-1,3-dithiane-1-oxide (6)
Thiophenol (0.4 mL g, 3.9 mmol) was added to sodium metal (89 mg, 3.9 mmol) in a small amount of THF under nitrogen, and the mixture was stirred for 10 min. The sodium phenylthiolate obtained was added to a solution of 4 (0.667 g, 3.9 mmol) in THF (10 mL), and the mixture was stirred for 12 h. Aqueous saturated NaCl solution (10 mL) was added, and the organic layer was separated. The aqueous layer was extracted using CHCl3 (3 × 20 mL), and the extracts and original organic layer were combined and dried (MgSO4). The solvent was removed under reduced pressure to give 6 as a mixture of two diastereoisomers in a ratio of 65:35. One diastereoisomer was separated (290 mg, 30%) by flash column chromatography (silica; EtOAc:Et2O = 1:10 by volume) as a yellow oil. 1H NMR (400 MHz; CDCl3) δ 7.75–7.60 (m, 2H), 7.36–7.29 (m, 3H), 5.09 (s, 1H), 3.20–2.88 (m, 3H), 2.49–2.16 (m, 3H). 13C NMR (125 MHz; CDCl3) δ 133.9, 132.8, 129.5, 128.8, 69.2, 47.3, 28.7, 24.2.
4.8. Reaction of 2-chloro-1,3-dithiane-1,3-dioxide (3) and trioctylborane
1-Octene (0.40 mL, 2.5 mmol) was added in a dropwise manner to a stirred solution (0°C) of borane (0.08 mL, 0.8 mmol; 10.0 M in Me2S) in THF (5 mL). The mixture was warmed up to 20°C and stirred for 1 h, and then cooled to –78°C. A solution of NaHMDS in THF (0.96 mL, 1.2 equiv.; 1.0 M) was added to a cooled (0°C) suspension of 3 (150 mg, 0.8 mmol) in THF (6 mL) and the mixture was cooled (–78°C). To that solution, tri-n-octylborane prepared earlier was transferred through a cannula. The mixture was stirred at –78°C for 3 h and allowed to warm up to 20°C over 1 h. A solution of aqueous NaOH (3.0 M, 10 mL) was added, followed by aqueous H2O2 (30%; 6 mL), and the mixture was stirred overnight. The organic layer was separated, followed by removal of solvent to give 1-octanol (230 mg, 72% of all octyl groups of tri-n-octylborane). The aqueous layer was acidified (HCl; 12 M) and extracted with DCM (3 × 20 mL). The organic layer was separated, dried (MgSO4) and the solvent was removed under reduced pressure to give 7 (64 mg, 50%) as a colorless oil. 1H NMR (500 MHz; CDCl3) δ 11.09 (br s, 1H), 2.34 (t, J = 7.5 Hz, 2H), 1.68–1.58 (app. quintet, J = 7.4 Hz, 2H), 1.40–1.17 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz; CDCl3) δ 180.6, 34.3, 31.9, 29.3, 29.2, 29.2, 24.8, 22.8, 14.2.
4.9. Reaction of 2-methoxy-1,3-dithiane-1-oxide (5) and trioctylborane
1-Octene (0.23 mL, 1.44 mmol) was added in a dropwise manner to a stirred solution of borane (48 µL, 0.48 mmol; 10.0 M in Me2S) in dry THF (5 mL). The mixture was warmed up to 20°C and stirred for 1 h. To the mixture, a solution of 5 (80 mg, 0.48 mmol) in THF (5 mL) was added. The mixture was cooled (–78°C) and n-BuLi in hexane (0.36 mL, 1.47 M, 0.53 mmol) was added dropwise. The mixture was stirred at –78°C for 1 h and allowed to warm up to 20°C and stirred for 1 h. The mixture was oxidized using an aqueous solution of NaOH (3.0 M, 10 mL), followed by aqueous H2O2 (30%, 6 mL). The mixture was stirred overnight and then saturated with NaCl. The crude product was extracted with CHCl3 (3 × 20 mL), and the organic layers were combined and dried (MgSO4). The solvents were removed under reduced pressure, and the residue obtained was purified by flash column chromatography (silica; EtOAc:Et2O = 4:96 by volume) to give 10 (16 mg, 13%) as a colorless solid, m.p. 48–49°C (lit [Citation41]. 48.5–49°C). 1H NMR (400 MHz; CDCl3) δ 2.38 (t, J = 7.5 Hz, 4H), 1.60–1.44 (m, 4H), 1.34–1.16 (m, 20H), 0.87 (t, J = 6.9 Hz, 6H). 13C NMR (125 MHz; CDCl3) δ 211.7, 43.0, 32.0, 29.5, 29.5, 29.3, 24.1, 22.8, 14.2.
4.10. Pummerer rearrangement in the reaction of 5 and trioctylborane
A solution of tri-n-octylborane (0.337 g, 0.96 mmol) in THF (5 mL), prepared as described above, was added to a solution of 5 (160 mg, 0.96 mmol) in THF (5 mL). The mixture was cooled to –78°C and n-BuLi (1.57 M in hexane; 0.72 mL, 1.13 mmol) was added in a dropwise manner. The solution was stirred at –78°C for 1 h and then allowed to warm up to 20°C. The mixture was cooled to 0°C, and a solution of TFAA (0.19 mL, 1.37 mmol) in DCM (5 mL) was added. The mixture was stirred at 0°C for 3 h and then warmed up to 20°C. NaOH (3.0 M, 10 mL) was added to the mixture, followed by aqueous H2O2 (30%, 6 mL), and the mixture was stirred overnight. The mixture was saturated with NaCl and extracted with CHCl3 (3 × 20 mL). The organic layers were combined, dried (MgSO4), and evaporated under reduced pressure to leave a colorless solid. The crude products were purified by flash column chromatography (silica; EtOAc:Et2O = 4:96 by volume) to give 10 (10 mg, 4%) as a colorless oil, 11 (22 mg, 6%) as a colorless solid, and 1-octanol (258 mg, 69% of all octyl groups of tri–n–octylborane) as a colorless liquid.
4.11. Synthesis of (3-(sec-butylsulfinyl)propyl)(methoxymethyl)sulfane (12)
A solution of sec-BuLi (1.4 M in hexane, 0.37 mL, 0.52 mmol) was added in a dropwise manner to a cold (–78°C) stirred solution of 5 (79 mg, 0.48 mmol) in dry THF (10 mL). The mixture was stirred for 15 min at –78°C and saturated aqueous NH4Cl solution (5 mL) was added. The mixture was warmed to 20°C and extracted with CHCl3 (3 × 10 mL). The combined extracts were dried (MgSO4) and evaporated under reduced pressure to give a mixture of the two diastereoisomers of 12 (55 mg, 49% yield) as a colorless oil. νmax (neat) 2964, 2926, 2875, 2802, 1423, 1055, 1029, 894, 749 cm–1. 1H NMR (500 MHz; CDCl3) δ 4.57 (s, 2H), 3.29 (s, 3H), 2.90–2.38 (m, 5H), 2.15–1.96 (m, 2H), 1.91–1.74 (m, 1H), 1.56–1.40 (m, 1H), 1.22, 1.14 (2d, each J = 6.9 Hz, total 3H), 1.03–0.96 (m, 3H). 13C NMR (125 MHz; CDCl3) δ 75.60, 75.58, 66.0, 57.3, 56.6, 56.0, 47.6, 46.9, 30.3, 23.9, 23.7, 23.3, 22.7, 15.4, 12.0, 11.5, 11.0, 10.9. EI-MS m/z (%) 224 (M+, 3%), 179 (M+ – OMe, 10), 163 (20), 148 (46), 107 (72). HRMS: Found for M+ (C9H20O2S2): 224.0899, calculated: 224.0905.
4.12. Synthesis of (3-(butylsulfinyl)propyl)(methoxymethyl)sulfane (13)
The procedure was identical to that used in Section 4.11 except that n-BuLi (0.33 mL, 1.6 M in hexane, 0.53 mmol) was used instead of sec-BuLi. Following work-up, 13 (45 mg, 40% yield) was obtained as a colorless oil. νmax (neat) 2927, 1550, 1055, 1026, 727 cm–1. 1H NMR (400 MHz; CDCl3) δ 4.62 (s, 2H), 3.33 (s, 3H), 2.56–2.84 (m, 6H), 2.10 (pentet, J = 7.1 Hz, 2H), 1.73 (app. pentet, J = 7.8 Hz, 2H), 1.43–1.54 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz; CDCl3) δ 75.6, 55.9, 52.4, 51.0, 30.2, 24.7, 23.1, 22.2, 13.8. EI-MS m/z (%) 224 (M+, 3%), 179 (M+ – OMe, 48), 163 (58), 148 (63), 107 (85). HRMS: Found for M+ (C9H20O2S2): 224.0899, calculated: 224.0905.
Acknowledgements
The authors thank Cardiff University and the Government of Iraq for their support. G. A. El-Hiti thanks the Researchers Supporting Project number (RSP-2021/404), King Saud University, Riyadh, Saudi Arabia. B. A. Saleh is grateful to the University of Basrah for supporting him during his Ph.D. study leave.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- He Z, Hu Y, Xia C, et al. Recent advances in the borylative transformation of carbonyl and carboxyl compounds. Org Biomol Chem. 2019;17:6317–6325.
- Fyfe JWB, Watson AJB. Recent developments in organoboron chemistry: old dogs, new tricks. Chem. 2017;3:31–55.
- Kaur P, Khatik GL, Nayak SK. A review on advances in organoborane-chemistry: versatile tool in asymmetric synthesis. Curr Org Synth. 2017;14:665–682.
- Smith K. Organoboron chemistry. In: Schlosser M, editor. Organometallics in synthesis: a manual. Chichester: Wiley; 2002. p. 465–533.
- Wang H, Jing C, Noble A, et al. Stereospecific 1,2-migrations of boronate complexes induced by electrophiles. Angew Chem Int Ed. 2020;59:16859–16872.
- Sandford C, Aggarwal VK. Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem Commun. 2017;53:5481–5494.
- Namirembe S, Morken JP. Reactions of organoboron compounds enabled by catalyst-promoted metalate shifts. Chem Soc Rev. 2019;48:3464–3474.
- Aggarwal VK, Fang GY, Ginesta X, et al. Toward an understanding of the factors responsible for the 1,2-migration of alkyl groups in borate complexes. Pure Appl Chem. 2006;78:215–229.
- Brown HC, Katz JJ, Carlson BA. Facile transfer of tertiary alkyl groups from boron to carbon in the base-induced reaction of α,α-dichloromethyl methyl ether with organoboranes containing tertiary alkyl groups. Novel route to highly hindered trialkylcarbinols involving exceptionally mild conditions. J Org Chem. 1973;38:3968–3970.
- Brown HC, Carlson BA. Fast base-induced reaction of α,α-dichloromethyl ether with organoboranes. New general route from organoboranes to the corresponding carbon structures. J Org Chem. 1973;38:2422–2424.
- Ncube S, Pelter A, Smith K. Improved synthesis of tertiary alcohols from reactions of organoboranes with 2-lithio-1,3-benzodithioles. Tetrahedron Lett. 1979;20:1895–1896.
- Ncube S, Pelter A, Smith K. Reactions of organoboranes and 2-lithio-2-alkyl-1,3-benzodithioles. A new, improved synthesis of ketones. Tetrahedron Lett. 1979;20:1893–1894.
- Hughes RJ, Ncube S, Pelter A, et al. Synthesis of secondary or tertiary alcohols by reactions of trialkylboranes with acyl carbanion equivalents. J Chem Soc Perkin Trans 1. 1977:1172–1176.
- Hughes RJ, Pelter A, Smith K, et al. Preparation of secondary alcohols by reaction of trialkylboranes with bis(phenylthio)methyl-lithium. Tetrahedron Lett. 1976;17:87–88.
- Hughes RJ, Pelter A, Smith K. Novel approach to the high yield synthesis of tertiary alcohols using organoboranes. J Chem Soc Chem Commun. 1974: 863https://doi.org/10.
- Podlech J. Stereoelectronic effects in α-carbanions of conformationally constrained sulfides, sulfoxides, and sulfones. J Phys Chem A. 2010;114:8480–8487
- Page PCB, McKenzie MJ, Allin SM, et al. Electrophilic amination of ketone enolates mediated by the DiTOX asymmetric building block: enantioselective formal synthesis of α-aminoacids. Tetrahedron. 2000;56:9683–9695.
- Page PCB, McKenzie MJ, Allin SM, et al. Enantioselective synthesis of α-methyl carboxylic acids using a DiTOX chiral auxiliary. Tetrahedron. 1997;53:13149–13164.
- Page PCB, Slawin AMZ, Westwood D, et al. Diastereoselective alkylation of ketone enolates using a 1,3-dithiane 1-oxide auxiliary. J Chem Soc Perkin Trans 1. 1989: 185–187.
- Carey FA, Dailey OD, Hernandez O. The stereoselectivity of reactions of electrophilic species with 2-lithio-1,3-dithiane 1-oxide. J Org Chem. 1976;41:3979–3983.
- Aggarwal VK, Boccardo G, Worrall JM J, et al. 2-Halogeno-1,3-dithiane 1,3-dioxide: a diastereoselective carbonyl anion equivalent in reactions with aldehydes. J Chem Soc Perkin Trans 1. 1997:11–20.
- Smith K, Saleh BA, Alshammari MB, et al. Studies on a catalytic version of the Matteson asymmetric homologation reaction. Org Biomol Chem. 2021;19:4279–4284.
- Smith K, Alshammari MB, El-Hiti GA. Unravelling factors affecting directed lithiation of acylaminoaromatics. Synthesis. 2018;50:3634–3652.
- Saleh BA, Smith K, Elliott MC, et al. Reactions of organoboranes with carbanions bearing three potential leaving groups: unusual processes, products and mechanisms. Tetrahedron. 2016;72:6914–6928.
- Jones DH, Smith K, Elliott MC, et al. Factors affecting reactions of trialkylcyanoborates with imidoyl chlorides/trifluoroacetic anhydride. Tetrahedron. 2015;71:6285–6289.
- Elliott MC, Smith K. Migratory aptitudes of alkyl groups on boron: a computational study of halomethyllithium-induced migration reactions. Organometallics. 2013;32:4878–4881.
- Elliott MC, Smith K, Jones DH, et al. Factors affecting migration of tertiary alkyl groups in reactions of alkylboronic esters with bromomethyllithium. J Org Chem. 2013;78:3057–3064.
- Smith K, Balakit AA, El-Hiti GA. Poly(propylene sulfide)–borane: reagent for organic synthesis. Tetrahedron. 2012;68:7834–7839.
- Smith K, Balakit AA, Pardasani RT, et al. New polymeric sulfide–borane complexes: convenient hydroborating and reducing reagents. J Sulfur Chem. 2011;32:287–295.
- Smith K, El-Hiti GA, Hegazy AS, et al. A simple and convenient high yielding synthesis of substituted isoindolines. Heterocycles. 2010;80:941–956.
- Smith K, El-Hiti GA, Hegazy AS. One-pot synthesis of substituted isoindolin-1-ones via lithiation and substitution of N'-benzyl-N,N-dimethylureas. Chem Commun. 2010;46:2790–2792.
- Aggarwal VK, Franklin RJ, Rice MJ. Highly stereoselective addition reactions of metallated trans-1,3-dithiane-1,3-dioxide to aldehydes. Tetrahedron Lett. 1991;32:7743–7746.
- Brown HC, Rogić MM, Rathke MW. Reaction of organoboranes with α-bromo ketones under the influence of potassium tert-butoxide in tetrahydrofuran. A new technique for the α-alkylation of ketones. J Am Chem Soc. 1968;90:6218–6219.
- Brown HC, Rogić MM, Rathke MW, et al. Stereochemistry of the carbonylation, carbethoxymethylation, and γ-propanalation reactions of organoboranes. Substitution reactions at carbon that proceed with retention of configuration. J Am Chem Soc. 1969;91:2150–2152.
- Bur SK, Padwa A. The Pummerer reaction: methodology and strategy for the synthesis of heterocyclic compounds. Chem Rev. 2004;104:2401–2432.
- Margot C, Rizzolio M, Schlosser M. 1,2-Elimination of alcohol from homoallyl ethers under the influence of mixed metal bases. Tetrahedron. 1990;46:2411–2424.
- Armarego WLF, Chai CL L. Purification of laboratory chemicals. Oxford: Butterworth-Heinemann; 2009.
- Watson SC, Eastham JF. Colored indicators for simple direct titration of magnesium and lithium reagents. J Organomet Chem. 1967;9:165–168.
- Carlson RM, Helquist PM. Synthesis and anionic properties of 1,3-dithiane 1-oxide. J Org Chem. 1968;33:2596–2598.
- Aggarwal VK, Davies IW, Franklin RJ, et al. X-ray crystal structure, equilibration studies and anion chemistry of trans-1,3-dithiane 1,3-dioxide. J Chem Soc Perkin Trans. 1991;1:662–664.
- Pelter A, Hutchings MG, Smith K. The chemistry of organoborates. Part III. Protonation of trialkylcyanoborates. J Chem Soc Perkin Trans 1. 1975: 142–145.