
ABSTRACT
Objective
The Aluetta™ reusable pen device and instructions for use (IFU) for growth hormone (r-hGH; Saizen®, Merck KGaA, Darmstadt, Germany) administration were tested for Human-Factors Usability, to ensure it could be used safely and effectively by the intended users in the intended use environment.
Research design and methods
Usability testing was conducted under simulated conditions in three groups of participants: pediatric or adult patients with growth hormone deficiency (GHD), participants without GHD, and healthcare professionals (HCPs). The testing comprised a 45-minute training session, a 2-hour testing session, and a participant-feedback session.
Results
Twenty-six participants completed the training session and performed all critical tasks related to the pen use across three scenarios. The most difficult tasks were related to the preparation, checking, and maintenance of the device; only 8% of use errors occurred during tasks related to the injection process. Eighty-five percent considered the pen safe and effective to use without further modifications and the training to be clear and effective.
Conclusions
The pen device and associated materials benefited from Human Factors Engineering throughout the development process. These evaluations show that patients and HCPs could safely and effectively use the pen device, and the IFU and training were clear and effective.
1. Introduction
Recombinant human growth hormone (r-hGH) is used in the treatment of several growth disorders in children [Citation1] and in adults with growth hormone deficiency (GHD) [Citation2]. The treatment regimen with r-hGH requires daily subcutaneous injections over a long period of time, which may not be conducive to maintaining good treatment adherence, particularly in children and adolescents [Citation3]. Poor adherence is associated with suboptimal clinical outcomes [Citation4–Citation7]. Several injection systems have been developed as alternatives to the traditional needle and syringe, with the aim of improving acceptability and ease of use. These include: syringes with hidden needles; disposable pre-filled injector pens; electronic auto-injectors; needle-free injectors; and reusable injector pens [Citation8,Citation9]. As GH injection devices have evolved, patients have expressed a preference for those that are easy-to-use, with features that reduce injection pain and offer minimal disruption to their daily routine. Furthermore, ongoing education on pen devices and injection technique, as well as ongoing support from healthcare professionals (HCPs), are likely to increase patient motivation to better adhere to therapy and therefore improve treatment outcomes [Citation8]. Moreover, improved awareness of the strengths and limitations of GH injection devices was identified as important for HCPs to guide families when selecting and using GH injection devices [Citation9].
The Aluetta™ pen device is a reusable, multi-dose injection pen for use with r-hGH (Somatropin [Saizen®], Merck KGaA, Darmstadt, Germany) [Citation10]. The new pen device was designed to provide an easy to use, lightweight, reusable, multi-dose injection device for patients injecting Saizen®. It comprises a multi-dose injection mechanism, a body constructed of aluminum, together with a multi-use cartridge system, a full cartridge-viewing window, a single dose-display window with a rotating dose-selection knob, and an injection button (; Figure S1). The device is available for 6, 12, and 20 mg presentations. Owing to the restrictions on the availability of the injection device (i.e., only through specialty pharmacies), all patients with GHD, caregivers, and healthcare providers are expected to be trained before using the pen device.
Figure 1. The three presentations of the pen device (blue: 6 mg; red: 12 mg; yellow: 20 mg).

For medical devices, the most important goal of the human factors/usability engineering process is to minimize the risks related to the use of the device. Therefore, applying Human Factors considerations early in the design process and then systematically throughout all development stages, with iterative improvements of the user interface is essential toward minimizing use errors. The Aluetta™ pen device, packaging, and instructions for use benefitted from the comprehensive application of Human Factors Engineering throughout the development process, in accordance with IEC62366-1 and IEC62366-2 [Citation11,Citation12] and the UK Medicines and Healthcare Products Regulatory Agency guidelines [Citation13]. The Human Factors Engineering comprised four formative evaluations and a summative (validation) usability test, guided by thorough use-related risk analysis. This process ensured that any risks related to the use of the device were identified and all changes made to mitigate these risks were evaluated to confirm that they were effective. All such errors were fed back into the product design, and the risk assessment process was repeated. As a result, the design of all parts of the device–user interface (including labels, instructions for use, and packaging) will have eliminated or reduced, as far as possible, any use errors that may cause harm.
As reported by Lange et al. in 2014, during the conduct of their late-stage formative usability study of a pen injector platform device, there are limited data available in the literature from formal usability studies of pen injection devices [Citation14]. Despite the paucity of such data, it does emphasize the importance of easy to understand instructions for use (IFU) and provide objective evidence that the intended use of a pen injection device has been met and can be safely and reliably used by the intended patient population.
To this end, the aim of this article is to document the results of the final human factors summative usability testing for the Aluetta™ pen device, performed in accordance with the relevant regulatory standards for usability/human factors engineering [Citation11,Citation12,Citation15]. This evaluation aimed to demonstrate that the pen device could be used safely and effectively by patients and HCPs to administer r-hGH under the expected use conditions.
2. Participants and methods
2.1. Participants and location
The summative human factors usability testing sessions took place at The Research House, London, UK. The study was conducted according to the European Pharmaceutical Market Research Association Code of Conduct [Citation16]. Institutional review board/ethics approval was not required because these were non-clinical, simulated-use studies; the summative usability testing was done under simulated-use conditions to enable a thorough assessment of the nature of the device and the IFU. All participants provided written informed consent before participation in any aspect of the study.
The participant sample comprised three groups: the pediatric group was comprised of injection-trained pediatric patients with GHD and representative pediatric participants without GHD (aged 13–17 years); the adult group was comprised of injection-trained adult patients with GHD (aged ≥ 18 years), representative adult participants without GHD (aged ≥ 18 years), and caregivers; and the HCP group comprised injection-trained HCPs.
2.2. Injection device, IFU, and packaging
During the summative human factors usability testing, participants interacted with the pen device, the IFU, and the packaging. The materials used during the summative study were representative of the final product but were blinded so that the name of the manufacturer, the brand name, and the drug name were not recognizable to the user (). The instructions to prepare the device and complete the injection, divided into 11 sections in the IFU, were: 1. Gather your supplies; 2. Select your pen injector and cartridge; 3. Inspect your cartridge; 4. Insert your cartridge; 5. Choose and prepare your injection site; 6. Attach your needle; 7. Prime your pen injector; 8. Dial your dose; 9. Inject your dose; 10. Remove and throw away your used needle and empty cartridge; 11. Cleaning and storage. The IFU contained the fake product name ‘Tatuela Pen’ and the pen device and the packaging were also modified with the fake product name. There were three variants of the injection device, designed for use with pre-filled Saizen® 6 mg, 12 mg, and 20 mg cartridges, respectively. The pen devices were packaged in their plastic storage box, within an outer cardboard box; likewise, the cartridges were contained within a plastic tray inside an outer cardboard box. To reflect real-world conditions, participants were provided with access to three needle types: Pencylcap™ (B Braun, Melsungen, Germany), AutoShield DUO™, (Becton Dickinson [BD], Franklin Lakes, NJ, USA), and one.click (EMD Serono, Inc., Rockland, MA, USA), allowing them to select their needle of choice. Injections were performed into simulated skin injection pads.
2.3. Training session
The training session was consistent with the training given in real-world situations. Each participant completed a 45-minute one-to-one training session with a nurse trainer. The trainer explained the content of the IFU, and two practical simulations were carried out: first, a simulated injection was performed by the trainer while the trainee observed; second, a simulated injection was performed by the trainee while the trainer observed. This was followed by discussion of any questions or concerns.
2.4. Testing sessions
The test sessions lasted for up to 2 hours and were conducted individually at least 1 day after the training session, to mirror the period that would elapse between training and first independent use of the pen device. The content of the sessions comprised use scenarios, knowledge tasks, and an interview; each session was recorded. These use scenarios were designed to compare the participants’ ability to prepare and deliver an injection with a single cartridge (scenario 1) or when an injection was split over two cartridges (scenario 2). These use scenarios also assessed the participants’ ability to prepare and deliver an injection using a single cartridge when the IFU was optional (scenario 1) or mandatory (scenario 3). Further details of the scenarios and knowledge tasks used during the testing session can be found in . The participants provided their feedback of each evaluation during an interview built around open-ended questions, with the aim of identifying the causes of any problem or error. Another part of the testing consisted of asking the participants to find and interpret important information in the IFU; for example, checking the pen and cartridge, selecting the appropriate injection site, and maintaining the pen.
Table 1. Use scenarios and knowledge tasks.
2.5. Definition of errors or potential errors
Errors or potential errors were recorded when a participant incorrectly performed or did not complete a task with the potential for harm to the patient or user (use error), when a participant nearly performed a task incorrectly but resolved the issue before any harm was done (close call), or when a participant struggled to some extent when completing a task but was able to complete the task (use difficulty). After the participant performed all use scenarios and knowledge tasks, and provided his/her impression of each use scenario and knowledge task, test personnel conducted a final, post-test interview. The interview included open-ended questions and focused on identifying the root causes of any use scenario or knowledge task failures, use errors, close calls, and difficulties that were not discussed earlier in the test session (e.g., during the post-use scenario interviews). The analysis of the use error’s root cause(s) was based on these observations and the professional judgment of the testers. Modifications to the IFU, pen injector, and packaging in response to the root causes, which were defined during the testing process as the fundamental reasons for the use errors, could be made to change performance and prevent an undesirable outcome.
3. Results
3.1. Participants
In total, 26 participants were included in the study (): eight in the pediatric group, nine in the adult group, and nine trained HCPs.
Table 2. Participant demographics.
3.2. Participant subjective feedback
Participants were asked to provide subjective feedback on the safety of the pen device, and the clarity of the IFU and the training. Twenty-two of the 26 participants (85%) considered the pen to be safe to use as it was presented, and four participants suggested some modifications. The participants’ suggestions were analyzed with a risk-based approach to determine if modification to the IFU, pen injector, and packaging were necessary. It was deemed that the device remained safe to use without suggestions for the pen injector and packaging being implemented.
Twelve of the 26 participants (46%) considered the IFU to be clear and effective as it was. Fourteen participants (54%) suggested changes to the text of the IFU (ten changes overall). After full assessment of the consequences of implementing the modification suggestions, one of these changes was implemented (a caution that it is not necessary to remove excess medication from the needle after priming). The remaining suggestions were considered either to have been already adequately addressed in the IFU, to introduce ambiguity to the document or risk to the users, or were not aligned with best practice guidelines.
Twenty-three of the 26 participants (88%) considered the training to be clear and effective. Three participants suggested to increase the length of the training session and to provide an online video of the pen instructions.
3.3. Use errors
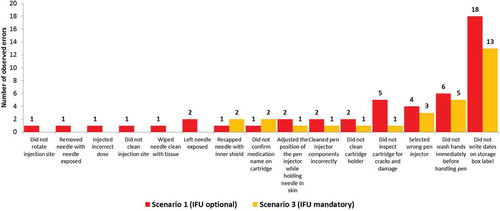
All twenty-six (100%) participants performed all critical tasks as part of the real-world scenarios. Successful task performance was generally high for all of the critical tasks. One or more overall use errors were reported for 16 tasks (). Grouping the use errors according to tasks shows that 87% of use errors occurred during the tasks related to the preparation, checking, and maintenance of the device, whereas the tasks relating to the injection process accounted for 8% of the use errors; the remaining 5% of use errors related to the removal/disposal of the needle. Most errors (40%) were made by the pediatric participants. Adult participants made 34% of errors and HCPs made 26% of errors (). There were several tasks related to sterility and aseptic procedures that all three user groups found more difficult to recall and perform: washing hands, writing the date of first use, cleaning the cartridge holder, cleaning the pen injector, rotating the injection site, checking the medication name on the label, and inspecting the cartridge for damage. Regarding injection with a single cartridge, when the participants were required to read and follow the IFU (scenario 3), the overall number of use errors dropped to 60% of the use errors recorded when the use of the IFU was optional (scenario 1) ().
Figure 2. Analysis of overall use errors (related to critical task) with potential for harm.
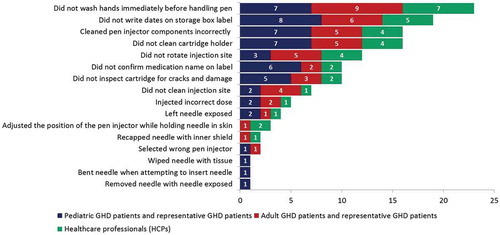
Figure 3. Total number of use errors on critical tasks per user group.
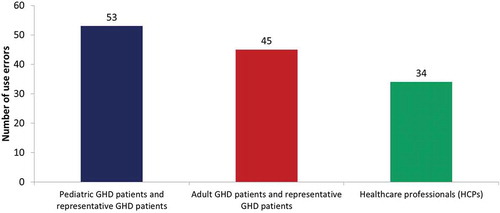
Figure 4. Number of observed errors with potential for harm in scenario 1 and scenario 3.
An assessment of the root causes and risk analysis were performed. Eighteen root causes were identified, leading to eight amendments to the IFU to mitigate some of the use errors by providing greater clarity, and without introducing any additional risks. All events associated with potential for harm that occurred during the summative usability testing were also assessed, along with the associated root causes, which were identified from the test participants’ comments, the observations of the test team, and follow-up human factors engineering analysis. On the basis of this analysis, the appropriate risk mitigations to minimize use errors and ensure the safe and effective use of the pen device were identified and implemented. Any risks associated with the pen device or IFU that remained after this thorough assessment were deemed as acceptably low. The amendments to the IFU resulting from this analysis are shown in and include an additional hand washing reminder and more explanatory illustrations. Clear reasons for not incorporating amendments to the IFU are also included, such as ‘instructions on the calculation of the dose are already included in the IFU’. If incidents were isolated, they were not deemed to be a risk and no change was made to the IFU. The modifications to the IFU were implemented with the aim to increase visibility or clarify wording. These modifications were rated as having a minor impact on the use of the device; therefore, implementation was not deemed to introduce any additional risks and further validation was not regarded as necessary.
Table 3. Analysis of the use errors, root causes and action taken.
3.4. Close calls relating to critical tasks
Five types of close calls were identified: 32% related to preparation of the skin, 31% related to preparing and checking the device, 24% related to maintenance of the device, 5% related to removal/disposal of the needle, 4% related to setting the dose, 3% related to the injection, and 1% related to priming the device before injection (Figure S2).
3.5. Use difficulties related to critical tasks
Three types of use difficulties related to critical tasks were identified: 57% were related to not checking the pen name and variant and cartridge and needle, 29% were difficulties removing the needle after the injection, and 14% were difficulties setting the appropriate dose by turning the dose knob.
3.6. Adverse events
Out of 26 participants, two needle-stick injuries occurred during the conduct of the study, both of which resolved without medical attention. During scenario 1, one participant in the adult group who was experienced in daily injections of r-hGH pricked her left index finger after pulling the needle cap off without untwisting the needle and brushed her hand against the exposed needle. The injury healed fully after 2 days. During scenario 2, one participant in the adult group who was inexperienced in daily injections of r-hGH pricked his left index finger while transferring the injector from the left to the right hand with the needle exposed. The individual did not continue with the simulated injections but completed the IFU interpretation tasks. The patient later confirmed by telephone that he was fine.
4. Discussion
In the current manuscript, we report the summative human factors testing of the new pen device. The new pen device incorporates several features that are designed to improve the user experience, such as an aluminum body, a multi-use cartridge system, cartridge viewing and dose display windows, a rotating dose selection knob, and an injection button. This evaluation showed that the pen device and associated materials could be used safely and effectively by the intended users, similar to the outcome reported after the functionality and ease of use testing of the pen device [Citation10]. The testing process presented here is consistent with other usability engineering assessments for pen devices [Citation17–Citation20]. These assessments are designed to ensure that, through an iterative refinement process (of which a validation study is the final phase), the pen injector and associated user materials can be used safely and effectively to perform critical tasks identified through the risk-management process and that the device training is effective and easy to follow. Devices that have poor user interfaces or instructions that are difficult to understand are more likely to be used incorrectly and deter patients from treatment, diagnosis, or monitoring; in some cases, they can lead to user harm or even death.
All new medical devices are required to undergo usability (human factors) engineering and risk assessment, which provide objective evidence that the device–user interface can be used safely by the intended users, in the intended use environment and for the intended use. The International Standard for usability testing of medical devices (IEC 62366 Part 1), which was first published in 2007 and updated in 2015, specifies the usability requirements for developing medical devices and provides the benchmark for compliance with regulatory requirements for the European Union [Citation11,Citation12] and United States markets [Citation21]. The results of human factors testing have led to high usability and acceptance of devices for the administration of a diverse range of treatments [Citation19,Citation22].
Using production equivalent versions of the pen devices, IFU, and packaging, as well as end-user training for the summative human factors testing, 26 participants performed all critical tasks. Most of the participants (85%) considered the pen to be safe and effective to use without any further modifications and also considered the training to be clear and effective. However, some improvements to the IFU were implemented to address, as far as possible, the root causes of the critical findings. These modifications to the IFU aimed to place important messages in more prominent positions and clarify the wording. Risk assessment shows that the changes did not introduce additional risks. The two adverse events reported out of 26 participants related to needle-stick injuries. These adverse events resolved without action and this type of injury would be a hazard for all injection devices that include needles.
Root causes of the main use errors could be attributed to five potential categories: clinical practice, the IFU and/or label, the multistep process of the injection, test artifacts, or negative transfer. In this assessment, the highest number of root causes, either in isolation or combination, were attributed to clinical practice, the multistep process, and test artifacts. Clinical practice relates to a task that is part of the injection process, independent of the design of a specific device (e.g., failure to wash hands before injecting is likely to occur, irrespective of the device being tested); multistep process refers to the complexity of the various tasks the users were expected to perform during the testing session, causing them to make errors or to have insufficient recall; and test artifacts relate to errors that are due to the effects of the simulated environment under which the tests were carried out, which may have led some participants to fail to take the same level of care and caution that they may exercise when preparing for and during the actual injection. Accordingly, many of the root causes were not specifically related to the specific injection device being tested, but would apply to any injection device, or there is a high probability they would not occur in real-world usage. As previously mentioned, the root causes attributable to omissions, misrepresentations, or unclear instructions in the IFU or label were investigated and changes were made where feasible. None of the root causes was thought to be caused by negative transfer, which is related to the previous experience of patients with other devices.
The use errors reported here mostly occurred during tasks related to the preparation, checking, and maintenance of the device (87%); only 8% of the use errors occurred during tasks related to the actual injection process and 5% of the use errors related to the removal/disposal of the needle. Furthermore, there was a decrease in the number of use errors from first use in scenario 1, when use of the IFU was optional, to the number of use errors in scenario 3, when the use of the IFU was mandatory. This shows that the IFU was effective for completing the tasks without error for the adult and pediatric participants.
There were some limitations to this summative human factors testing study, which could be applicable to all studies of this type. While the training environment was highly reflective of the real-world training environment, it was carried out in an office-based environment; furthermore, the injection procedure was a simulated use scenario, with participants performing injections into injection pads placed on the part of their body where they would inject in real life, rather than into actual patients.
5. Conclusions
This summative human factors evaluation demonstrated that the pen device could be used safely and effectively by patients, nurses, and caregivers to administer r-hGH under the expected use conditions according to the regulatory standards for usability/human factors engineering. This assessment also supported the effectiveness of the IFU, with users given the opportunity to assess potential risks and to mitigate any risks by suggesting changes to the wording of the IFU; such changes were minimal, supporting the effectiveness of the original document.
Following the human factors validations, the pen device was deemed as safe and effective for use by the intended user populations in the intended use environments.
Author contributions
In accordance with the stipulations set out by the ICMJE, we confirm that all authors made substantial contributions to the concept and design, or analysis and interpretation of data, as well as either drafting the manuscript or revising it critically for important intellectual content. Furthermore, all authors contributed to the development of the instructions for use, the test plan, and the test report. In addition, all authors provided final approval of the manuscript to be published and agree to be accountable for all aspects of the work.
Declaration of interest
SE Warren is an employee of EMD Serono, Inc., a business of Merck KGaA, Darmstadt, Germany. At the time of the study, A Cachemaille and S Moss were employees of Ares Trading S.A., an affiliate of Merck KGaA, Darmstadt, Germany. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Data availability
Data sharing should be done in accordance with the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Pharmaceutical Research and Manufacturers of America’s (PhRMA) Principles for Responsible Clinical Trial Data Sharing. Merck KGaA, Darmstadt, Germany, believes that as a biopharmaceutical company, the sharing of information related to company sponsored clinical trials is central to our mission. The sharing of clinical trial Information enables the medical and scientific community to further develop the medical and scientific knowledge base and permits the public to make informed healthcare decisions. However, because information from company sponsored clinical trials may include confidential personal information and proprietary company information, Merck must ensure that information is provided only in response to legitimate scientific and medical requests and that information disseminated outside of the company properly protects all confidential. All clinical trial information must further be provided only in accordance with applicable laws and codes. Merck will share anonymized patient level, and study level data and redacted clinical study reports from clinical trials in patients with qualified scientific and medical researchers, upon researcher request via the Merck website portal (https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html), as necessary for conducting legitimate research. More information for researchers can be found on this website. Evaluation of the Research Proposal, as well as the qualifications and experience of the Lead Researcher and Research Team, will be conducted by appropriate and qualified person or board. The researcher must enter into a Data Sharing Agreement (‘DSA’) with Merck. The standard DSA which must be entered into is posted on Merck’s website. Merck uploads the data to Cloud-Based Data Sharing Analytical Solution where researchers can conduct the approved analysis. A request form is available on the Merck website portal (https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html). A point of contact for information regarding data requests is Sascha-Marc Seidl ([email protected]).
Acknowledgments
The authors acknowledge medical writing assistance from Steven Goodrick of inScience Communications, Springer Healthcare, Ltd, London UK and acknowledge Emergo by UL which completed the testing in the study. The authors would also like to thank all of the participants who took part in the study.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- National Institute for Health and Care Excellence. Human growth hormone (somatropin) for the treatment of growth failure in children: technology appraisal guidance [TA188]. London: NICE; 2010.
- National Institute for Health and Care Excellence. Human growth hormone (somatropin) in adults with growth hormone deficiency: technology appraisal guidance [TA64]. London: NICE; 2003.
- Kapoor RR, Burke SA, Sparrow SE, et al. Monitoring of concordance in growth hormone therapy. Arch Dis Child. 2008 Feb;93(2):147–148.
- Bagnasco F, Di Iorgi N, Roveda A, et al. Prevalence and correlates of adherence in children and adolescents treated with growth hormone: a multicenter Italian study [Multicenter study]. Endocr Pract. 2017 Aug;23(8):929–941.
- Cutfield WS, Derraik JG, Gunn AJ, et al. Non-compliance with growth hormone treatment in children is common and impairs linear growth [Research support, non-U.S. Gov’t]. PLoS One. 2011 Jan 31;6(1):e16223.
- Sabaté E. Adherence to long-term therapies: evidence for action. Geneva: World Health Organization; 2003.
- Fisher BG, Acerini CL. Understanding the growth hormone therapy adherence paradigm: a systematic review [Review]. Horm Res Paediatr. 2013;79(4):189–196.
- Rohrer TR, Horikawa R, Kappelgaard AM. Growth hormone delivery devices: current features and potential for enhanced treatment adherence [Review Research Support, Non-U.S. Gov’t]. Expert Opin Drug Deliv. 2017 Nov;14(11):1253–1264.
- Raimer-Hall D, Shea HC. Evolution of growth hormone devices: matching devices with patients [Comparative Study Research Support, Non-U.S. Gov’t Review]. Pediatr Nurs. 2015 Mar-Apr;41(2):72–77.
- Sauer M, Abbotts C. A new pen device for injection of recombinant human growth hormone: a convenience, functionality and usability evaluation study. Patient Prefer Adherence. 2018;12:27–34.
- International Organization for Standardization. IEC 62366-1:2015 medical devices - part 1: application of usability engineering to medical devices. Geneva: International Organization for Standardization; 2015.
- International Organization for Standardization. IEC 62366-2:2016 medical devices - part 2: guidance on the applicability of usability engineering to medical devices. Geneva: International Organization for Standardization; 2016.
- UK Medicines and Healthcare Products Regulatory Agency. Human factors and usablity engineering - guidance for medical devices including drug-device combination products. London: MHRA; 2017.
- Lange J, Richard P, Bradley N. Usability of devices for self-injection: results of a formative study on a new disposable pen injector. Med Devices (Auckl). 2014;7:195–203.
- European Medicines Agency. Good practice guide on risk minimisation and prevention of medication errors. Eur Med Agency. 2015. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/good-practice-guide-risk-minimisation-prevention-medication-errors_en.pdf
- European Pharmaceutical Market Research Association. EphMRA code of conduct. Bromley, Kent (UK): European Pharmaceutical Market Research Association; 2017.
- Mahony M, Dwyer A, Barkume R, et al. US human factors engineering evaluation of an updated follitropin alfa pen injector (GONAL-f® RFF Redi-ject®) and instructions for use. Expert Opin Drug Deliv. 2018 Jan;15(1):5–15.
- Jeannerot F, Studeli T, Gunther-LaVergne L, et al. Usability engineering study in the European Union of a redesigned follitropin alfa pen injector for infertility treatment. Expert Opin Drug Deliv. 2016 Sep;13(9):1221–1229.
- Travanty MN, Calawa B, Shalaby WS, et al. Development and usability of a new subcutaneous auto-injector device to administer hydroxyprogesterone caproate to reduce the risk of recurrent preterm birth. Med Devices (Auckl). 2018;11:241–252.
- Fujioka K, Sparre T, Sun LY, et al. Usability of the novel liraglutide 3.0 mg pen injector among overweight or obese adult patients with or without prior injection experience. J Diabetes Sci Technol. 2015 July 16;10(1):164–174.
- US Food and Drug Administration. Guidance for industry and food and drug administration staff: applying human factors and usability engineering to medical devices. Rockville, MA, USA: FDA; 2016.
- Wilcox SB, Drucker D. Implications of the new food and drug administration draft guidance on human factors engineering for diabetes device manufacturers. J Diabetes Sci Technol. 2012 Mar 1;6(2):231–235.