
ABSTRACT
Introduction
The development of long-acting injectables (LAIs) for protein and peptide therapeutics has been a key challenge over the last 20 years. If these molecules offer advantages due to their high specificity and selectivity, their controlled release may confer several additional benefits in terms of extended half-life, local delivery, and patient compliance.
Area covered
This manuscript aims to give an overview of peptide and protein-based LAIs from an industrial perspective, describing both approved and promising technologies (with exceptions of protein engineering strategies and devices), their advantages and potential improvements to aid their access to the market.
Expert opinion
Many LAIs have been developed for peptides, with formulations on the market for several decades. On the contrary, LAIs for proteins are still far from the market and issues related to manufacturing and sterilization of these products still need to be overcome. In situ forming depots (ISFDs), whose simple manufacturing conditions and easy administration procedures (without reconstitution) are strong advantages, appear as one of the most promising technologies for the delivery of these molecules. In this regard, the approval of ELIGARD® in the early 2000ʹs (which still requires a complex reconstitution process), paved the way for the development of second-generation, ready-to-use ISFD technologies like BEPO® and FluidCrystal®.
1. Introduction
The market of therapeutic biologics keeps growing and expanding, with novel treatments being approved over the last years and an increasing number of products in pharmaceutical development pipelines [Citation1,Citation2]. Between 2015 and 2018 for example, 30% of all drugs approved by the FDA were biologics, and the global peptide market is expected to reach $50 billion in 2027, doubling its value compared to only three years ago [Citation3,Citation4]. Although small molecules still represent a large proportion of global sales (about 90%), the clear increase of proteins and peptides approved is due to their high efficacy and lower toxicity compared to small molecules [Citation4,Citation5].
The growing interest around the use of peptide therapeutics is well documented by the huge number of molecules in active clinical development, estimated to be around 170, covering a wide range of applications. Considering only approved molecules, areas such as urology, respiratory, and cardiovascular diseases, pain, oncology, metabolic disorders, and infections are most commonly associated to peptide drugs treatments [Citation6]. Regarding protein therapeutics, monoclonal antibody-based therapies have had incredible success over the last decades, due to the high specificity and usually long half-life of these molecules. Monoclonal antibodies found many applications for indications such as cancer (e.g. Trastuzumab or Bevacizumab) or immune diseases (e.g. Adalimumab or Infliximab). However, many other protein therapeutics like hormones, cytokines, growth factors, and enzymes, were approved in the past and are currently used for the treatment of many diseases (e.g. multiple sclerosis, Gaucher’s disease or rheumatoid arthritis) [Citation7,Citation8].
Despite the specificity and selectivity of protein and peptide therapeutics, there is a common interest for improving their pharmacokinetic properties, both by using molecular biology (e.g. fusion proteins or other protein engineering strategies) and drug delivery systems [Citation9,Citation10]. The delivery strategies are case-dependent and rely on several factors like the properties of the molecules, the targeted indication, the type of delivery system, the route of administration and even patient-related factors. However, considering LAIs, defined as injectable formulations which deliver therapeutic active molecules and extend their half-life from days to months, some common, recognized advantages can be drawn. In fact, longer half-lives avoid the need for frequent administrations, a clear advantage in terms of patient compliance and reducing potential side effects like injection-site reactions. LAI technologies also allow the control of the burst release phase, avoiding potential adverse reactions associated to high initial drug blood concentrations [Citation11–14].
In this regard, several long-acting technologies were approved in the past decades for the delivery of peptides, mostly formulated with polylactic acid (PLA) and poly(lactic-co-glycolic) acid (PLGA), both of which were approved in the ’70s for pharmaceutical applications [Citation15]. These polyesters did not achieve the same successful delivery of proteins, for which no products can yet be found on the market.
In contrast, lipidic delivery systems, especially liposomes, found a wide application for the release of small molecules, with several products approved in the past (e.g. Doxil®, Exparel® or Ambisome®) [Citation16]. However, no liposomes were marketed for the long acting delivery of therapeutic proteins and peptides [Citation17].
The goal of this review is to describe, from an industrial perspective, the LAI landscape for peptides and proteins and the associated technologies, which are broadly presented in and . Examples of marketed products, those in clinical trials or relevant preclinical works are presented, while considering that fusion proteins/peptides and delivery devices are out of the scope of this work.
Figure 1. Potential controlled-release technologies for the delivery of peptides and proteins. The technologies already marketed are presented in the figure underlined and in bold.
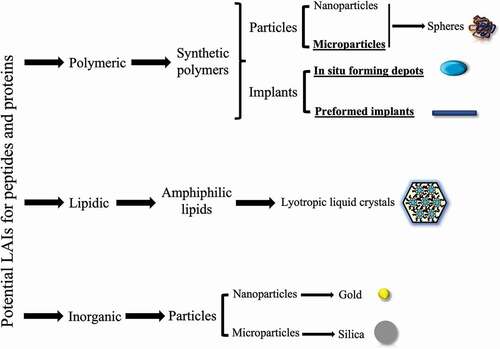
Table 1. General overview of the existing LAI technologies used for the delivery of proteins and peptides.
An analysis of the difficulties behind the development of LAIs for the delivery of biotherapeutics and of the future perspectives associated to these technologies is also given in . However, considering that challenges and limitations are strictly related to parameters like the route of administration, the type of moleculemolecules, the materials used and the application for which the LAIs are developed, aims at giving only a broad overview of these topics. On the contrary, an analysis of the main strengths and weaknesses of approved peptide LAIs, as well as the key hindrances for the approval of protein LAIs, with the identification of the principal bottlenecks, is developed in the last sections of this manuscript.
Table 2. Challenges, limitations, and future perspectives of LAIs for proteins and peptides. The first line of the table presents general limitations and perspectives, common to nanoparticles, microparticles, ISFDs and solid implants. Challenges and perspectives specific to each one of these systems is then presented in the subsequent lines of the table.
2. Long-acting injectables for peptides
Therapeutic peptides must often be administered chronically, over significant periods of time. Due to their very low permeation through membranes, they often show low bioavailability when administered by oral or transdermal routes. For these reasons, they must be administered parenterally and hence have been obvious candidates for the development of LAIs due to their short half-life, rapid clearance, and intrinsic instability. Moreover, peptides are generally very potent molecules, which translates to relatively low daily doses (usually ≤1 mg), a strong advantage when designing weekly or monthly formulations for therapeutic indications requiring chronic administration. So far, only eleven LAIs for peptides have been approved by the FDA, which underlines the complexity of making such drug products (). All of them are administered subcutaneously or intramuscularly and the majority are made using PLA or PLGA-based microspheres or implants.
Table 3. Long-acting injectables for peptides currently commercialized. m: month(s); w: week(s).
An overview of the main strategies from an industrial perspective, for the manufacturing of these delivery systems, with a short description of the processes and their limitations is given in . An extensive description of all these products is then given below.
Table 4. Overview of the principal formulation strategies for the manufacturing of PLA/PLGA microspheres, solid implants, and in situ forming depots.
2.1. PLGA/PLA-based peptide-containing microspheres
2.1.1. Critical parameters governing peptide release and microsphere properties
Whatever the adopted manufacturing scheme, it has been well established that the drug release from polyester-based microspheres presents three phases: i) an initial burst that can account for the release of a significant fraction of the drug cargo followed by ii) a lag phase or induction time, days to weeks, where a minimal drug release is observed, that could result in drug exposure below a therapeutic threshold, before iii) a continuous release of the molecule [Citation38]. The initial burst is often associated to the release of the drug molecules located close to the surface of the microspheres. This burst may hence raise toxicity concerns, since the drug loading is intended to provide the appropriate dose for the expected duration of the release, which might be up to several months. Hence, for example a burst release of 10% for a 1-month formulation may correspond to the recommended dose for 3 days, which might be higher than the toxicity threshold. The intermediate lag phase appears after the burst period, when a low release occurs due to the intrinsic properties of the polymeric matrix (i.e. the absence of pores). The third phase corresponds to the continuous release of the API and is closely related to polymer degradation [Citation39,Citation40].
PLA polymers or PLGA copolymers are key excipients of such drug products as they will determine their sustained release features. For example, the key role of using PLGA instead of PLA for the development of faster-releasing products, due to its more rapid degradation, was already understood in the ’80s and led to the development of Lupron Depot® [Citation41–43]. Several studies have also established the key physico-chemical characteristics for these types of products. For instance, a low polymer porosity correlates well with a low initial burst in vitro, as seen with Bydureon® and Sandostatin® LAR [Citation14,Citation44,Citation45]. Other characteristics of the polymer such as, molecular weight, blockiness (the presence of potential GA clusters in PLGA polyester in contrast to a random copolymer structure) or architecture (linear vs branched), may influence the peptide release profile [Citation46–48].
2.1.2. Current limitations of microsphere-based products (acylation, RTU, manufacturing)
As peptides are intrinsically fragile molecules, the question of their stability during encapsulation, manufacturing processes, and subsequent release, is key. In fact, peptide primary amines may covalently bind to the polyester backbone, forming acylated peptide forms [Citation49,Citation50]. This has been demonstrated with Sandostatin® LAR, for which 30% to 40% of the released octreotide is acylated. The problem may be related to the use of acetate as counterion, which favors the formation of negatively charged polymer degradation species which, in turn, react with the positively charged octreotide [Citation44]. The use of alternative salts (e.g. octreotide pamoate) or the addition of divalent cations like Ca2+ or Mn2+ may limit the peptide sorption to the polyester backbone, avoiding acylation [Citation51–53]. Other impurities than acylated ones, like oxidized and deamidated species, were also observed on the exenatide extracted from Bydureon® [Citation14]. Indeed, peptide instability may be one of the main reasons behind the low bioavailability (ca. 25%) observed for this product compared to the Byetta®, which is an immediate exenatide release form.
Another limitation of microsphere-based products is due to their manufacturing process (refer to ), which is rather complex and could explain the lack of emergence of generic products through the 505(j) FDA pathway [Citation54]. In fact, given the multi-step and multi-parameter characteristics of the procedures, the number of critical variables to consider is impressive (e.g. type of organic solvent, process temperature, extraction volume, PVA concentration, etc.). In this perspective, the production of microspheres would undoubtedly benefit from adopting a Quality by Design (QbD) approach, which, from the critical material attributes and critical process parameters, would allow the definition of Critical Quality Attributes (CQAs) for the final drug product [Citation55].
LAIs are sterile products, meaning that their manufacturing process needs to include a sterilization step, either through sterile filtration or terminal sterilization. However, terminal sterilization is not always feasible due to the potential instability of the formulation (both peptide and polymers), as the sterilization process is usually based on gamma-irradiation or dry heat treatment. Dry heat would induce polymer and/or peptide degradation while gamma sterilization can induce polymer degradation [Citation56]. Therefore, the whole process must be implemented in an aseptic environment, which increases the complexity and the costs for the development and commercial manufacturing of such LAIs.
Another potentially troublesome feature of some peptide-loaded microsphere-based LAIs is their very low initial burst. This low initial peptide serum concentration, remaining below the therapeutic threshold for a significant period of time, translates into a longer onset time, which could require initial supplementation with the immediate release form, like in the case of Sandostatin® LAR. On the other hand, this feature can be exploited for APIs whose elevated serum concentration are associated with side effects, as for exenatide; for example, Bydureon® releases exenatide over 8 to 10 weeks, leading to a progressive increase of the exenatide serum concentration and the establishment of a steady state regimen after seven to eight successive weekly injections [Citation57,Citation58].
An additional limitation to the widespread use of microsphere-based products may also be their frequently tedious mode of reconstitution and administration. Most of the commercialized products must be reconstituted extemporaneously by mixing the dried microspheres with a diluent and most of them must be administered under the supervision of a healthcare professional. Even though Lupron Depot® is commercialized as a prefilled dual-chamber syringe that can be stored at room temperature, it still requires a 9-step process before administration. The reconstitution sequence for the Sandostatin® LAR is even less convenient, with a 30-min to 1-h incubation at room temperature prior to reconstitution, the observance of a 5 min saturation time after addition of the diluent injection into the vial containing the microspheres and a 30s shaking of the vial with the risk of aspirating and incomplete dose into the syringe. The different successive presentations of Bydureon® from a vial to a dual-chamber pen, which still required a multi-step reconstitution and had to be shaken ‘hard like you would shake a bottle of oil-and-vinegar salad dressing,’ to the current ready-to-use (RTU) BCise® pen, illustrate well how a complex drug product can be tailored to a much more convenient one for self-administration [Citation59].
2.2. Bioresorbable solid implants for peptides
2.2.1. Presentation of the dosage form and of currently commercialized drug products
A class of LAIs for peptides that has also been very successful in terms of regulatory approval and commercialization is that of solid implants. Due to the limitations in terms of implant size and the need of high polymer content for controlling the drug delivery, one of the major drawbacks of such products is the relatively low API loading. The three drug products described in (Zoladex®, Suprefact® and Scenesse®) are all PLGA rods prepared by hot-melt extrusion (HME) (refer to ) [Citation60]. Once implanted, the release of the peptide is controlled by the rate of polymer degradation and drug diffusion through the polymer matrix. Similarly to microspheres, the release profile generally shows the same three phases (initial burst, lag-phase and faster release linked to polymer erosion) which can be tuned by using polymers with different properties [Citation61].
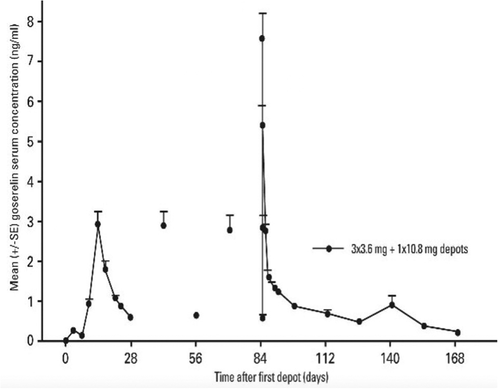
Interesting information can be gained having a closer look to these products. The oldest product, Zoladex®, a ready-to-use PLGA implant of goserelin acetate for the treatment of prostate cancer and the management of endometriosis or advanced breast cancer, was approved by FDA in 1989 as a 4-week product (3.6 mg peptide dose). A 12-week version (10.8 mg dose) was later approved in 1996. The diameter of the 12-week implant is 1.5 mm, while the 4-week one is 1.1 mm, a difference which is reflected by the different needle size necessary for their administration (i.e. 16 G for the 12-week and 14 G for the 4-week). Both Zoladex® versions come as prefilled syringes and the amount of PLGA in both products is similar, around 15 mg. However, even if information regarding the polymer molecular weight is rather difficult to retrieve, differences in the nature of PLGA can be found, with a LA:GA ratio of 50:50 and 95:5 for the 4-week and 12-week products, respectively [Citation62]. Although the goserelin amino acid sequence does not contain primary amines that could be the locus of acylation reactions, the FDA specifies that up to 12% of goserelin-related substances can be retrieved from implants. The release profile is strikingly different between both implants. The 4-week version presents a very slow initial release with a Tmax comprised between 12 and 15 days (Cmax around 3 ng/mL), with declining concentrations over the next 15 days. In contrast, the Tmax for the 12-week product is only 2 hours (Cmax around 9 ng/mL), with peptide concentrations decreasing rapidly over the next 4 days to stabilize thereafter ().
Figure 2. Mean goserelin serum concentration in prostate cancer patients (n = 41) after the dosing of three ZOLADEX® 3.6 mg implants (days 0, 28 and 56) followed by one ZOLADEX® 10.8 mg implant (day 84). Concentration profile has been taken from NDA 020578/S-032.
The strategy to design the 2-month and 3-month buserelin implants (Suprefact® Depot) has been different. As goserelin and buserelin are GnRH agonist approved for the treatment of advanced hormone-dependent prostate cancer. Both products present the same drug loading of 20% (w/w) for both the 6.3 and 9.45 mg doses and use PLGA with the same LA:GA ratio of 75:25. The 2-month and 3-month implants integrate, respectively, two or three identical rods packaged in a ready-to-use applicator equipped with a 14 G needle for subcutaneous injection. The 2-month product reaches its Cmax (around 2 ng/mL) a few hours after administration, followed by a slight decline over the next 60 days. It was estimated that around 90% of the buserelin cargo had been released after the two-month period. In the same manner, a Cmax of 8 ng/mL was reached 4 hours after injection of the 3-month implant, followed by a concentration declining over 2 weeks to reach steady state concentrations, maintained until the end of the treatment period.
The most recent product is Scenesse®, an implant of Afamelanotide, a synthetic 13 amino acids peptide analog of the a-melanocyte hormone, that is used to prevent skin damage in people with erythropoietic protoporphyria. The drug product comes as a solid rod implant (1.7 cm long, 1.45 mm diameter) containing 16 mg of peptide and 15 to 19 mg of PLGA. Although the product must be administered every 2 months, the implant releases most of the peptide within the first 48 hours (Tmax at 36 h), with 90% of the initial cargo being released at day 5 [Citation63]. Despite the rapid peptide release, it has a prolonged effect on the melanin density in tissues that was at its peak at day 28 and still noticeable up to 60 days. The implant was fully resorbed 50 to 60 days after implantation, suggesting that the PLGA presents a GA content higher than 20%. The implantation procedure is not easy as the implant must be taken out a sealed glass vial stored at 4°C and pushed into a cannula that has been previously placed into the subcutaneous layer. The implant is then simply pushed down the full length of the cannula with a stylet.
The description of the different commercialized implant products shows that this formulation approach is attractive, especially regarding the relatively good stability of this dosage form. Additionally, it is possible to modulate to some extent the peptide release by tuning the drug loading and/or the PLGA nature. However, the main limitations are related to the mode of administration that requires large needles that are painful and affect the adherence of patients to the treatment [Citation64].
2.3. In situ forming depot technologies
2.3.1. Presentation of currently commercialized drug products
Despite the advantages of in situ forming depot (ISFD) technologies and the simple mechanism of implant formation, only two peptide-loaded products (Eligard® and Camcevi®) based on this technology are currently commercialized. They both use the Atrigel® technology that relies on PLGA polymers that are solubilized in a biocompatible solvent, N-methyl-2-pyrrolidone (NMP) [Citation65]. However, thanks to a counterion exchange strategy substituting mesylate with acetate that offers a better stability profile, Camcevi® comes as a ready-to-use drug product [Citation66]. One feature of ISFD technologies, especially for those relying on a solvent exchange mechanism, is the presence of an initial burst. This is often related to the highly hydrophilic nature of proteins and peptides, which rapidly diffuse into the surrounding environment. However, advantages like higher loadings compared to microspheres or solid implants, the simple sterilization process (usually by sterile filtration) and the possibility of being removed if necessary, make them appealing for the development of future products [Citation67].
Having a closer look at the different dosage forms of Eligard® provides us with interesting information about the key parameters that impact the formulation properties (see ). The product is commercialized as two syringes stored at 4°C, one holding the PLGA polymer-containing vehicle and the other one the leuprolide acetate powder. After 30 min at room temperature, the two syringes can be connected through a luer-lock system and the vehicle is injected into the syringe containing the powder. Correct mixing is ensured by a 45 second push-pull sequence between the two syringes. shows that to increase the release duration, the lactic acid content in the PLGA copolymer was increased as well as the polymer content in the PLGA vehicle. The injection volume increase was limited by incrementing the peptide loading from 3% (w/w) to 12% (w/w), ending up with a suspension instead of a solution for the most concentrated 6-month product, which explains the change in needle size. Polymer content and composition are key variables for modulating the release kinetics using this technology. This was also recently demonstrated with our proprietary BEPO® technology [Citation51,Citation68].
Table 5. Composition of the different Eligard® formulations from the product monograph.
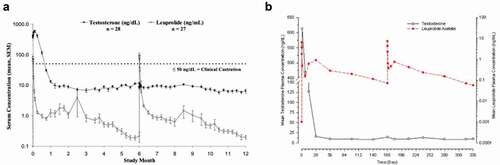
As previously described, the burst control is still the main issue of ISFDs. In this regard, it is interesting to compare Lupron Depot® to Eligard®, as both integrate the same dose of leuprolide acetate to achieve the same release duration. It is important to note that for this indication, an initial burst is not contra-indicated. The GnRH agonist will initially induce a flare response leading to a transient increase in testosterone levels. However, continuous administration leads to receptor internalization and to a hypogonadism state with testosterone concentrations below castration levels. A head-to-head comparison was made by injecting the two 1-month versions of the products in 16 patients per treatment group [Citation69]. Surprisingly, the Cmax concentration was 30% lower for the ISFD product than for the microsphere-based one (19 ng/mL versus 27 ng/mL) with similar Tmax of 1 to 2 hours. Eligard® also displayed a longer release duration (50 days versus 30 days) and a better bioavailability. However, when looking at the two 6-month versions, the burst release is much more pronounced for Eligard® than for Lupron Depot® (Cmax of 80 ng/mL instead of 7 ng/mL). The decline in concentration was faster and subsequent leuprolide acetate serum levels were lower for Lupron Depot® than for Eligard® but both products were able to maintain testosterone below castration levels [Citation70,Citation71] (). It has to be noted that the highest Cmax value is elicited by the 4-month 30 mg Eligard® product with a mean concentration of 150 ng/mL and that even a value of 212 ng/mL was observed when the 6-month Eligard® product was administered to a pediatric population for treatment of precocious puberty [Citation72].
Figure 3. Pharmacokinetic (leuprolide acetate concentration) and pharmacodynamic (testosterone level) response after two successive administrations of the 6-month version of the Eligard® (a) or Lupron Depot® (b) products. Both graphs have been taken from the FDA approval packages.
A similar 10-fold difference in Cmax and similar Cmin values was also observed when comparing the intramuscular Sandostatin® LAR microsphere-based product to the subcutaneous one, CAM2029 developed using the FluidCrystal® technology (refer to ) [Citation73]. Still, as discussed in the publication, the Cmax value of CAM2029 was considered acceptable in terms of toxicity as it was equivalent or even below the octreotide concentration reached after the injection of the highest dosage of the immediate release form.
3. Long-acting injectables for proteins
Despite 50 years of efforts and many papers published, no long-acting product for proteins, based on the use of drug delivery systems is available on the market.
This section of the review aims at explaining the main reasons for the failure of controlling protein delivery and elucidates the key challenges for the design of delivery systems for these molecules. A final focus on the existing technologies which have been approved, withdrawn from the market or are currently in clinical trials, and their applications will be given.
3.1. Proteins: structural considerations and post-translational modifications
Proteins are large macromolecules, ranging from few hundred to thousands of Dalton, whose three-dimensional organization is characterized by secondary, tertiary, or quaternary structures (the so-called higher order structures (HOSs)) distinctive of each protein and responsible for their bioactivity. The loss or even the alteration of HOSs during the production of the formulation or during the storage period may dramatically impact the bioactivity of biomolecules and even increase their immunogenic potential [Citation74]. This makes the development of formulation processes complicated, since they must not alter the HOSs of the molecules. Considering PLGA microparticles for example, they are often not suitable for the encapsulation and release of proteins, both for manufacturing reasons, which will be discussed later, and physico-chemical limitations [Citation75]. More precisely, in PLGA-based formulations the release of molecules is usually driven by the degradation rate of the polymers, at least after the burst release period. However, accumulation of acidic degradation products within the polymer matrix may decrease the local pH, affecting protein structure and potentially causing further degradation [Citation76,Citation77]. Given the strict correlation between protein structure and function, even slight modifications of HOSs may cause a loss of activity, compromising the efficacy of the product. In addition, the misfolding of proteins can also be a concern in terms of patient safety, since it may lead to aggregation, and aggregate-driven immunogenic reactions may occur [Citation78].
However, conformational concerns are not the only ones. Post-translational modifications (e.g. variation in glycosylation patterns, oxidation, and deamidation), sensitive to environmental factors like variations in temperature and pH for example, must be monitored during the entire process of formulation development and storage [Citation79].
3.2. Development of formulations for proteins: challenges, limitations, and scale-up considerations
3.2.1. Manufacturing
The manufacturing strategy is obviously related to the formulation type and process chosen. Considering the wide use of PLGA microparticles and implants, a focus is made on these technologies. In general, the same procedures and process limitations related to the production of peptide-loaded LAIs can also be proposed for the encapsulation of proteins (refer to ). However, being structurally more complicated molecules, their stability is highly impacted by the formulation strategy, a fact which further complicates the development of LAIs for protein therapeutics. Particular attention must be given to the encapsulation of proteins into microparticles by processes which uses organic solvents (e.g. double emulsion – solvent extraction or coacervation), which are among the most commonly used for peptides. A major limitation relies on the presence of water/organic solvent interfaces (both at lab and industrial scale), to which proteins tend to adsorb, leading to denaturation and aggregation [Citation80]. Stabilizers like surfactants or sugars can be added to the formulation to preserve protein structures as surfactants outcompete protein molecules for hydrophobic surfaces while sugars like cyclodextrins, sucrose, or trehalose, may form protective complexes [Citation81,Citation82].
In the case of implants produced by hot melt extrusion (HME), the impact of high temperatures and shear stresses is even larger for proteins than it is for peptides, which do not have any tertiary or quaternary structure. Excipients like sugars or poly(ethylene-glycol)s, which shield the proteins from the melted polymer, may increase their stability, but the HME procedure is still far from optimal [Citation83].
ISFD are probably a good alternative for overcoming the limitations of microparticles and preformed implants, since high temperatures and the use of harsh solvents are avoided. However, solvents like N-methyl-2-pyrrolidone (NMP) or dimethyl sulfoxide (DMSO) are often used and may induce protein denaturation [Citation84]. A valuable alternative was found in the use of small chain triglycerides like triacetin or tripropionin. For example, the substitution of DMSO with tripropionin allowed the successful formulation and half-life extension of a fully functional bi-specific antibody for treating prostate cancer [Citation85]. A PLGA/triacetin system also allowed in vitro sustained release of a Fab fragment to be achieved [Citation86].
However, regardless of the formulation approach chosen, mechanical stresses are the most common ones when scaling-up protein-containing formulations. In fact, classical formulation processes usually mix molecules and polymers that are then stirred in reactors. This exposes the molecules to shear stresses and air–water interfaces, leading to denaturation and aggregation. In this regard, homogeneous stirring, which avoids bubbles and the adsorption of molecules to surfaces, is key to deliver high-quality products to the market [Citation55,Citation87,Citation88].
3.2.2. Storage stability
The poor stability of protein LAIs is typically one of the key issues to overcome when developing drug products. Temperature, humidity, and protein concentration of the drug product can influence the physico-chemical properties of both the delivery system and the active ingredient [Citation89]. It is well known that the presence of water molecules may favor protein aggregation and cleavage, especially if the proteins are encapsulated in PLGA systems, where the hydrolytic degradation of the polymer backbone may induce the formation of a local micro-acidic environment [Citation90]. To circumvent this issue, many researchers have suggested the use of alkaline excipients to avoid polymer degradation and favor the preservation of protein structures [Citation91,Citation92]. Nevertheless, developing ready-to-use formulations for these types of molecules is complex, and industries turned their strategies to the manufacturing of dried products. Moisture is a major enemy as the presence of water may lead to plasticizing effects, which contribute to protein aggregation and degradation [Citation93,Citation94]. For these reasons, storage temperature plays a central role for the delivery of good-quality and safe products to the market. Products stable at room temperature are ideal in terms of logistics, availability for developing countries and costs. For instance, the World Health Organization estimated that the cold supply chain represents 20% of the total vaccines cost [Citation95]. However, biologics, i.e. proteins or peptides, are usually not stable at room temperature and storage at 4°C or below is often necessary [Citation96]. Besides temperature and pH, light, and the presence of contaminants and leachables coming from the manufacturing process or storage materials may be an issue [Citation97,Citation98].
3.2.3. Sterilization
Sterilization strategies are product dependent and differ based on the type of protein and formulation. However, some key points can be highlighted considering dried formulations of PLGA microparticles, implants, and ISFDs. As for peptides, sterilization by filtration is not possible with microparticle-based products but can be envisioned with ISFDs, a fact which further reinforces the interest around the use of these types of formulations. However, special attention should be paid when scaling-up filtration methods: during cross-flow filtration, for example, proteins may be exposed to high shear rates while passing through membranes, leading to potential denaturation. The passage through pumps and valves may also cause microcavitation, leading to the formation of bubbles, which, upon collapse, create microscopic regions at high local temperature and pressure which may contribute to the generation of hydrogen and hydroxyl radicals and lead to the formation of protein aggregates and particles [Citation87,Citation88].
If peptide-based products are usually sterilized by gamma irradiation and heat sterilization, those processes cannot be used with proteins due to their fragile nature (i.e. protein degradation or modifications may occur). In this regard, aseptic manufacturing conditions should be used, which increases the complexity of the manufacturing process and the price of the final products [Citation99]. Regardless of the formulation type and technology used, a key step remains the aseptic drying of the final product, a procedure carried out by spray-drying or freeze-drying. However, aseptic spray drying processes are still uncommon and difficult to achieve, with a limited number of contract manufacturing organizations offering this service [Citation100,Citation101]. On the contrary, freeze-drying is a well-established industrial process to produce sterile dried cakes made under good manufacturing practice (GMP) conditions [Citation100].
3.2.4. Immunogenicity: a perspective on the gaps between academy and industry
As previously explained, a key aspect of the development of protein formulations is the structural stability of the molecules throughout the steps of the production process, from manufacturing to sterilization and further storage. In fact, the unfolding of proteins may lead to the formation of aggregates, often designated as the main culprits for inducing immunogenic responses [Citation102]. Moreover, the materials used to manufacture the delivery systems may also cause immunogenicity by themselves. For instance, polyethylene glycols (PEGs), often used in the development of biopharmaceuticals and drug delivery systems, may elicit an immune response, leading to adverse reactions. The immunogenicity of PEGylated drugs or drug delivery systems depends on several factors, among them, chain length, branching, and type of terminal end-group [Citation103].
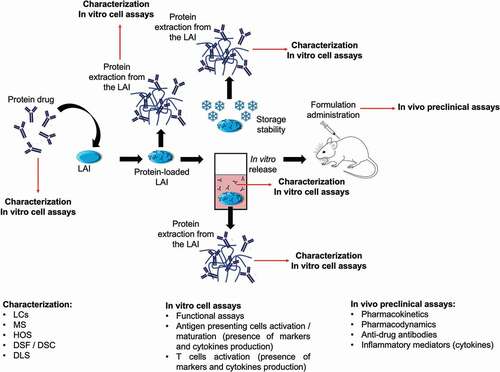
For this purpose, the FDA has defined some guidelines for assessing the immunogenicity risk during the clinical phases of product development and suggested the development of anti-drug antibody assays. The final goal is to identify the mechanisms which trigger the immune responses and to define potential immune response mitigation strategies (i.e. premedications, desensitization, or immune tolerance induction procedures) which might reduce or avoid the adverse reactions [Citation104]. However, it is important to assess and predict potential immune reactions in a preclinical context when possible. Here lies one of the principal reasons explaining the difference between the high number of papers published on drug delivery systems for proteins and the low number of products entering a clinical development. Often these publications do not focus enough on the characterization of the molecules at the different steps of the formulation development, when released or when associated to the delivery systems [Citation105,Citation106]. A very good package of analyses is given in the work of Fayd’herbe De Maudave et al., where several chromatographic techniques (size exclusion, reverse phase, and ionic exchange), mass spectrometry, differential scanning fluorimetry, and functional assays were used to study the structure and function of a formulated monoclonal antibody through an ISFD technology [Citation107]. Nevertheless, on top of these analyses, determination of the potential alteration of the protein’s high-order structures and the potential presence of subvisible particles by, for example, light obscuration or micro-flow imaging, should also be performed. Characterized samples should then be used to quantify and understand the protein-driven immune response, if any. Studying the whole mechanisms that support the immunogenic response is a challenge, both in vitro and, especially in vivo, where the use of animal models is often not representative of a human immunogenic response (i.e. species-dependent immunogenicity) [Citation108]. However, systematic studies regarding the role of antigen-presenting cells and on the activity of B and T cells can be and should be introduced, as suggested by several authors [Citation109–111]. A schematic representation of the approach to be used for developing long acting injectables for proteins, considering immune system activation, is given in .
Figure 4. Example of an experiment aiming to fully characterize the protein at the different stages of the formulation development and evaluating the immunogenicity of the protein in vitro and in vivo. Briefly, the protein should be characterized in its native state and after each formulation step thereafter. Each step is characterized by different stresses that may impact the structure of the protein with potential consequences in terms of immunogenicity. The in vivo preclinical assays remain fundamental to gain a deeper understanding of the mechanisms around immunogenic responses. LC: liquid chromatography; MS: mass spectrometry; HOS: high order structures; DSF: differential scanning fluorimetry; DSC: differential scanning calorimetry; DLS: dynamic light scattering.
3.2.5. In vitro – in vivo correlation (IVIVC): a clear limitation for the formulation of proteins
Despite years of efforts, few successful examples of functional IVIVC models can be found for peptide/protein LAIs. If the typical parameters to consider while designing models for small molecules are pKa, drug permeability, pH of the environment, and partition coefficient, dealing with proteins makes the modeling significantly more complicated [Citation112]. Proteins are large dynamic molecules, whose properties depend on several factors like pH, temperature, environment, and surrounding molecules (e.g. ions, fatty acids, sugars, etc.). The presence of a delivery system which can alter the conformation of the proteins and, as consequence, their release kinetics, further complicate the situation. For instance, Olthof et al. tried to correlate the in vivo release of bone morphogenetic factor – 2 from several composites (i.e. oligo(poly(ethylene glycol) fumarate hydrogels)) to the one obtained with classical in vitro set-ups (i.e. immersion of the hydrogels in different media). A poor IVIVC was observed, both in term of timings and amount of protein released. They related the poor prediction of the model to the complexity of the composites, from which drug release is driven by different mechanisms like polymer erosion, diffusion, desorption, and ion exchange and to the necessity of strictly mimicking the in vivo environment [Citation113].
A review of Jain et al. is also very interesting, since they highlighted the importance of the biological environment, especially the formation of protein corona, for defining the fate of drug-loaded nanoparticles. They described how classical in vitro set-ups do not take into consideration-specific tissue related and biological responses, such as drug interaction with cell receptors, inflammatory response, or adsorption to tissues [Citation114].
The review of Zolnik and Burgess is also a comprehensive overview of the parameters to consider for obtaining satisfactory IVIVC with different drug delivery systems. They suggested the inclusion of immune response – related factors into the definition of the in vitro models (e.g. production of anti-drug antibodies), as it may affect protein release and subsequent pharmacokinetics in vivo [Citation115].
3.3. Long-acting injectables for proteins: industrial perspective of products and clinical trials
As explained in the previous sections, there is no long-acting injectable product for proteins approved (see ). Despite the success of PLA and PLGA injectables for peptides, the same approach has been quite disappointing in the protein field. So far, the only approved product has been Nutropin Depot®, soon withdrawn from the market due to several formulation issues. In fact, the product was highly viscous, requiring large administration volumes with large and long needles (21 gauge, 1.5”) and frequent injections were necessary to achieve a therapeutic effect [Citation116].
Table 6. Long-acting injectable technologies for proteins. Approved products or relevant technologies in clinical or preclinical evaluation are reported in the table.
Nevertheless, MedinCell S.A. and Innocore Pharmaceuticals are two of the companies actively developing polyester-based formulations for the delivery of proteins. The BEPO® technology developed by MedinCell, an in situ forming depot technology which relies on the use of PEG-PLA copolymers, was used to release the bispecific antibody BiJ591 for 21 days in vivo after a subcutaneous administration to mice. It was shown that the burst release of the protein strongly depends on the type of copolymers used in the formulation [Citation85]. The same technology was used to deliver monoclonal antibodies in the intra-articular cavity of mice joints, with promising results for the development of potential treatments for osteoarthritis [Citation107]. On its side, Innocore Pharmaceuticals developed PCL-PEG-PCL-b-PLA multi-block random copolymer microparticles that were used to encapsulate pPB-HSA, a platelet derived growth factor-recognizing moieties (pPB) linked to human serum albumin (HAS). The usual half-life of the protein after intravenous injection is 45 minutes, while, using the Innocore technology, it was possible to quantify the biomolecule in the serum at least 7 days after the subcutaneous injection of the microparticles in mice [Citation117]. A similar technology named SynBiosys®, made of poly(caprolactone), or poly(lactic) acid, and PEG, was also developed by Innocore [Citation118,Citation119]. It is an in situ forming depot technology that was used to deliver several types of model proteins, such as insulin, lysozyme, carbonic anhydrase, and albumin [Citation119]. At the present time, only in vitro results were published regarding this technology.
Another company active in the field of protein delivery is DelSiTech, which develops formulations using silica microparticles dispersed in silica hydrogels. This system was used to encapsulate a model antibody, MR1, whose release was studied both in vitro and in vivo, upon subcutaneous injection in mice. Pharmacokinetic results clearly show the extended release of the antibody (up to 62 days) compared to the free antibody control, for which serum concentrations were undetectable already at day 20 [Citation120].
also summarizes some of the clinical trials of different long-acting technologies conducted during the last decade (taken from https://clinicaltrials.gov). An interesting work used gold nanoparticles (CYT-6091 – Aurimune™) for the delivery of TNF-α to advanced solid tumors. The association of the protein to nanoparticles confer stealth properties to the molecule, limiting its phagocytic clearance by the reticuloendothelial system. A phase I trial to test side effects and best doses after intravenous administration was carried out. No relevant side effects were measured and the association of the protein to the particles extended the protein half-life by up to 5 times (130 minutes vs 28 minutes) when compared to the free TNF-α, favoring its accumulation into the tumor 24 hours after treatment [Citation121]. Despite the good results, no recent updates were found on this topic (i.e. the last update date in 2012 – ClinicalTrials.gov identifier: NCT00356980). However, CYT-6091 can still be found in the pipeline of CYTIMMUNE as ‘phase 2 – ready’ [Citation122].
Other interesting technologies which were promising for the delivery of proteins but did not move further after the initial clinical trials are Medusa® and Locteron®.
Medusa® is a 10–50 nm nanoparticle suspension made of poly L-glutamate grafted with α-tocopherol. The formulation was used to encapsulate several proteins, among them IL-2, for the treatment of renal carcinoma (Phase I/II) and INF- α2b for hepatitis C (HCV) treatment (Phase I/II). Compared to Proleukin® (the commercial version of IL-2), Medusa® allowed the reduction of the Cmax by a factor 2 and extended the Tmax up to 48 hours (instead of the typical 2–4 hours). In the case of HCV, the Medusa® technology reduced the Cmax compared to Viraferon® by a factor of nine, extending the release of the protein up to 7 days and drastically reducing the side effects of the drug [Citation123]. However, even in this promising case, no further studies were found regarding these products.
Locteron™ is a controlled release formulation using microparticles that was designed for the delivery of a recombinant form of interferon-α (interferon-α 2b, produced by Biolex Therapeutic) in combination with oral ribavirin, to HCV patients. The delivery system, PolyActive®, made of poly(ether ester) multiblock copolymers based on poly(ethylene glycol) and poly(butylene terephalate), was produced by a company called OctoPlus N.V [Citation124]. A phase I/II study was started in 2008, comparing safety, tolerability, pharmacokinetics, and viral kinetics of interferon loaded Locteron™ (subcutaneous administration every 2 weeks), with PegIntron®, a commercial formulation (weekly injection) [Citation125]. It was shown that Loctaron™ treatment induced less frequent and severe side effects than PegIntron® and that anti-viral effects were comparable for both treatments [Citation126]. Despite the positive results and the will of testing the formulation in a Phase III study, the project did not move forward, probably due to the approval of a new treatment for HCV [Citation127].
Another interesting technology is Biochaperone®, produced by Adocia, a delivery system for several types of proteins. Biochaperone® forms complexes with proteins, protecting them from enzymatic degradation, enhancing their bioavailability, and stabilizing them during storage [Citation128]. The company is focusing on the treatment of diabetes and related disorders with several products in different phases of clinical trials [Citation129]. They also worked on the delivery of platelet-derived growth factor – BB (PDGF – BB) for treating chronic diabetic foot ulcers. Phase II studies showed positive results, with enhanced protein efficacy and reduced administration frequency when using the Biochaperone® technology compared to the commercial Regranex®. However, the company shut down the development of the product due to Phase III failure [Citation130].
4. Systemic and local delivery of proteins and peptides using LAIs
While systemic delivery of biological therapeutics by a controlled release system prolongs the plasmatic circulation of drugs, it is often difficult for those molecules to reach high-localized concentrations at the target site. In this sense, local delivery ensures high drug concentrations at the site of administration while minimizing off-target toxicity and immunogenicity [Citation131].
Nowadays, some of the most well-established local routes of administration include topical, subgingival, intra-articular (IA), intra-tumoral (IT), and intravitreal (IVI), the three latter ones are often used for the delivery of biological therapeutics.
4.1. Intra-articular (IA) delivery of biologics
Intra-articular (IA) administration is often used to treat joint-related diseases, like, for example, osteoarthritis (OA), for which there are still high unmet medical needs [Citation132]. In fact, when administered systemically, drug molecules reach the synovial compartment by vascular diffusion. However, the diffusion of large macromolecules like proteins is impeded due to their size and for this reason, IA administration is preferred. Nevertheless, these molecules are removed rapidly from the synovial fluid by lymphatic drainage and LAIs are necessary to ensure their long-term presence in the joint [Citation133]. So far, no long-acting IA delivery treatment using proteins or peptides is approved. Nonetheless, we recently obtained promising data in pre-clinical studies using our ISFD technology BEPO® to encapsulate a model monoclonal antibody, Rituximab [Citation134]. Despite the abundance of preclinical studies, no LAI for biologics has succeeded in advancing into clinical phases. One potential reason is that since IA injections are quite invasive and can only be performed by rheumatologists, it is widely acknowledged that a minimum treatment interval of 3 months should be considered, which is difficult to achieve. Additionally, alternative strategies like intra-articular injections of platelet-rich plasma and mesenchymal stem cells have been developed, limiting the interest around the development of IA LAIs [Citation135].
4.2. Intra-tumoral (IT) administration
The interest around local treatment of malignancies is demonstrated for example by several clinical trials around IT injection of ipilimumab, conducted in the past 10 years [Citation136–138].
The IT administration of biologics may help to overcome the typical limitations imposed by the tumor micro-environment, poorly permeable, and highly acidic, when anti-cancer agents are administered systemically [Citation139,Citation140]. Additionally, IT LAIs are also necessary to avoid frequent administrations, often not manageable given the location of certain tumors (e.g. brain, lung, liver, etc.) and the need for surgery [Citation141,Citation142]. Although many IT anti-cancer biological agents have progressed into clinical phases, their related long-acting formulations have not made much progress, mainly due to limitations like tumor heterogeneity and accessibility [Citation143]. A successful example is G100, a potent TLR4 agonist from Immune Design administered using an oil-in-water emulsion technology, which has recently completed a phase II study. G100 was shown to induce an anti-tumor immune response and tumor regression in patients with Merkel cell carcinoma [Citation141].
4.3. Intravitreal (IVI) administration
The intravitreal (IVI) administration route has been used to deliver many types of medications into the vitreous cavity. However, so far, the only biologics administered intravitreally are VEGF inhibitors (anti-VEGF antibodies and fusion proteins) [Citation144].
IVI injections are often uncomfortable and usually require one injection per month. Therefore, LAI formulations that could increase the dosing interval would be highly beneficial to patients. However, the main limitations for the development of IVI LAIs are the need for low viscosity formulations, administered by a limited injection volume (ideally 0.1 mL) [Citation145]. For instance, Kodiak Sciences developed KSI-301, an anti-VEGF antibody biopolymer conjugate, which has recently advanced into clinical phase III [Citation146]. A good product safety profile was demonstrated and compared to the non-conjugated aflibercept, it provided better efficacy and longer exposure.
5. Conclusion
The high specificity and selectivity of biologics make these molecules incredibly appealing for the development of novel therapeutic treatments. However, the development of LAIs for proteins and peptides has been limited by their intrinsic fragility. If several LAIs for peptides have been successfully commercialized during the last 20 years, the same cannot be said for proteins, whose formulation manufacturing and stability limitations still need to be overcome.
Polyester-based microparticles and preformed implants are currently the most commonly used technologies for peptide delivery, due to their ease of development and production from an industrial perspective. In situ forming depot LAI technologies are extremely appealing for the delivery of biologics, due to their relatively mild formulation conditions and the possibility for being injected locally. However, further investigation and development is still required.
6. Expert opinion
Peptide and protein therapeutics are extremely potent and specific. For these reasons, pharmaceutical companies have made significant efforts to develop products based on these molecules over the last 20 years. Despite their potential, difficulties related to formulation development and administration have limited their entrance into the market, especially regarding their LAI forms. In fact, biotherapeutic molecules often show:
- a low chemical and physical stability in physiological conditions, including in gastro-intestinal fluids,
- a low permeability through cells and membranes, which strongly affect their bioavailability when administered using non-parenteral routes of administration.
- a short half-life (with exceptions for monoclonal antibodies).
The oral delivery of biopharmaceuticals may appear to be a breakthrough for patients. The possibility to administer proteins and peptides without the need of injections, often uncomfortable and risky in term of patient’s safety, is extremely appealing. However, despite the attractiveness of the oral delivery route, the highly acidic environment of the stomach and the presence of enzymes, as well as the low permeability across the intestinal epithelium still hinder the development of oral products for biopharmaceuticals, with bioavailabilities ranging from 0.1% to 1% for peptides [Citation147].
Although many issues still need to be overcome, the interest of companies around this topic is strong, and is proven by the recent FDA approval of two peptides for oral administration, namely semaglutide (Rybelsus®) and octreotide (Mycapssa®). However, despite the presence of permeation enhancers, which are expected to improve the permeability of the epithelial membrane, the overall oral bioavailability for these molecules is still lower than 1% [Citation148]. This means that the manufacturing capacity and the properties of orally administered peptides must be significantly improved. Many efforts have been made to develop novel oral drug delivery technologies (e.g. self-nanoemulsifying drug delivery systems, novel permeation enhancers, silica particles, scaffold, pills, etc.), and many of them are currently in clinical trials [Citation147]. However, adapting the industrial manufacturing system to these potentially new products still requires time and investment, with strong potential impact on the cost of goods.
A further problem associated with the oral delivery of biopharmaceuticals is related to the variable inter- and intra-patient responses. As a consequence, there is a need for very specific daily administration schedules to avoid food effect (a fact which impact the adherence of patients to these treatments) [Citation149].
Bearing all this in mind, the development of long-acting formulations of peptides and proteins becomes key for the treatment of chronic diseases. LAI formulations avoid repeated (daily) oral administrations of the drugs, limiting variations in term of drug concentration in the blood stream and improving patients’ compliance in term of administration schedule. The shift from the use of implants and microparticles to in situ forming depots represent a further advantage while thinking to administration by classical injections. Many industries are in fact focusing on the development of low viscous, ready-to-use, self-administrable ISFD formulations, injected through thin needles (i.e. 25–27 gauge), which are well tolerated and not painful.
If many LAI drug products for peptides have been successfully commercialized during the last 20 years, the same cannot be stated for biologics. For these large molecules, stability during manufacturing, storage, and delivery are still challenges that need to be overcome. New formulation technologies, which avoid conditions where the protein is exposed to and may adsorb at interfaces, leading to denaturation, loss of bioactivity, and formation of aggregates, need to be developed. In situ forming depot technologies represent once again a first breakthrough for LAIs, due to their relatively mild formulation conditions and their potential for local injection. Most likely, there will be an additional need for solvent-free formulation technologies operating under mild conditions, e.g. processes using supercritical fluids [Citation150].
Concerns regarding the use of polyester-based materials should be also discussed. Polyesters may not be the most appropriate polymers for the formulation of LAIs for proteins. In fact, stability issues during storage and delivery, related to the acidic microenvironment generated by the degradation of the polyesters chains leading to protein degradation and aggregation, have often been reported. To this end, it is important to develop new polymeric scaffolds, as well as in vitro and in vivo assays which bridge structural modification of proteins to potential immunogenic responses. The production of anti-drug antibodies for example, often in response to protein aggregates, may reduce the pharmacokinetic performance of the administered molecules, compromising the benefits associated with LAI-based therapies. It is hence advisable to characterize both the structural stability of the delivered drug and potential immune responses that may occur, ideally by using human-derived materials (i.e. human serum and immune cells), and to collect as much data as possible during the first phases of the clinical trials.
Nevertheless, the development of new biomaterials which favor the stability of biologics and extend the half-lives of rapidly cleared molecules like hormones, cytokines, and engineered antibodies will likely need to be developed. This is expected to be an area of intense research activity in the coming years.
Regarding peptides, polyester-based microparticles and preformed implants are the most common technologies used for LAI formulations and probably the easiest ones to develop from an industrial perspective, given the already existing expertise and know-how in various pharmaceutical companies. However, these technologies still show strong limitations related to the manufacturing processes, which often require heating or the use of toxic solvents, reconstitution before administration (e.g. for microparticles) and the use of large Gauge needles (e.g. for preformed implants). The former may lead to reconstitution errors, needle sticks and, sometimes, clogging, while the latter to problems in terms of patient compliance, as previously discussed. The first generation of ISFD technologies like e.g. Atrigel® technology, addressed some of these concerns, but still requires a very complex reconstitution procedure which involves at least five successive steps and a mixing procedure in a push-pull syringe system.
The key challenge for these type of formulations remains the preparation of stable, ready-to-use liquid products, ideally in pre-filled syringes that can be administered through fine needles without any reconstitution. The second generation of ISFDs, based on technologies like FluidCrystal® or BEPO®, seem very promising in this perspective and may allow the development of improved LAI formulations of peptides for clinical applications. In addition, solvent-free technologies are also expected to provide great opportunities for the development of improved LAI formulations [Citation151,Citation152].
Injectable peptide formulations should remain the predominant and preferred products on the market over the next 5–10 years, considering that improved ready-to-use LAI formulations are developed and filed. This should be a strong driver for the further development of activities in this field. Products for alternatives routes of administration will surely be developed to compete with the parenteral routes, likely for some specific peptides for which oral administration may confer a significant advantage. This is the case for peptides targeting the liver or other organs in the gastro-entero-pancreatic environment. However, this will require the development and approval of much more efficient permeation enhancers than the present ones, involving for example functionalized nanocarriers actively targeting receptors at the surface of membrane cells, or smart micro-needle-based oral devices [Citation153].
In this regard, the development of clinically approved active targeting delivery systems may represent an important step-forward for the treatment of many cancers as well. The capacity to direct active molecules to tumors (or to a specific set of tumors cells) and to favor their permeation in the tumor mass (or in the cytoplasm of cells), would reduce side effects, enhancing the potency of those therapies. The use of carriers made by different materials and able to associate molecules with different properties, would also favor the development of combination therapies. However, despite the huge number of preclinical studies, no products based on a co-delivery strategy have yet been approved, probably due to the complexity of these systems, the lack of regulatory guidelines and the necessity of adapting pharmaceutical industries to the preparation of these new systems.
In contrast, a growing interest around the use of microneedle technologies (usually not injectable) for protein and peptide delivery was observed in the last years. The main advantage of these devices relies on their low invasiveness and capacity to penetrate the epidermis at a fixed depth, favoring the absorption of macromolecules through capillaries and the lymphatic system [Citation154]. The properties of microneedles can be tuned using different biocompatible or biodegradable materials, which allow the formation of hollow, swellable, or thermoresponsive structures. However, the manufacturing of microneedles is still expensive and requires specific fabrication methods. Many companies, for example NanoPass Technologies Ltd. or Zosano Pharma are active in the field, and several products can be found in clinical trials. Despite many preclinical studies claiming the successful use of microneedles for achieving long-lasting delivery of biotherapeutics, clinically relevant technologies are not often associated to a sustained, long acting delivery and, on the contrary, they are designed to promote the quick absorption of these molecules [Citation155–158].
Article highlights
LAIs for peptides and biologics reduce the frequency of administrations and may improve the pharmacokinetics of these molecules, and hence their safety and efficacy profiles.
The most commonly used technologies, which have led to approved products, are microparticles and preformed implants, commercialized for the delivery of several peptides.
There is still room for improving the currently available technologies, by developing liquid, stable formulations that are ready-to-use and easier to administer.
The main reason for the limited success of LAIs for proteins is related to the complexity and fragility of these molecules, which show poor chemical and physical stability during the manufacturing and sterilization processes used for preparing LAIs at industrial scale.
There is a general interest for developing in situ forming depot technologies, especially regarding the possibility of delivering therapeutic macromolecules locally.
Declaration of interest
All the authors are employees of MedinCell S.A. and shareholders of the company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Lagassé HAD, Alexaki A, Simhadri VL, et al. Recent advances in (therapeutic protein) drug development. F1000Res. 2017;6:113.
- What Are “Biologics” Questions and Answers | FDA [Internet]. cited 2021 Sep 9. Available from: https://www.fda.gov/about-fda/center-biologics-evaluation-and-research-cber/what-are-biologics-questions-and-answers
- Anselmo AC, Gokarn Y, Mitragotri S. Non-invasive delivery strategies for biologics. Nat Rev Drug Discov. 2019;18(1):19–40.
- D’Aloisio V, Dognini P, Hutcheon GA, et al. PepTherDia: database and structural composition analysis of approved peptide therapeutics and diagnostics. Drug Discov Today. 2021;26(6):1409–1419.
- Makurvet FD. Biologics vs. small molecules: drug costs and patient access. Med Drug Discov. 2021;9:100075.
- Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther. 2022;7(1):48.
- Reynolds T, de Zafra C, Kim A, et al. Overview of biopharmaceuticals and comparison with small-molecule drug development. Nonclinical dev nov biol biosimilars vaccines spec biol [Internet]. Elsevier; 2013 [cited 2022 Jun 3]. p. 3–33. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780123948106000010
- Dimitrov DS. Therapeutic Proteins. In: Voynov V, Caravella JA, editors. Ther Proteins Internet]. Totowa (NJ): Humana Press; 2012, 1–26. [cited 2022 Jun 3]. Available from: http://link.springer.com/10.1007/978-1-61779-921-1_1
- Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29(4):215–239.
- Vaishya R, Khurana V, Patel S, et al. Long-term delivery of protein therapeutics. Expert Opin Drug Deliv. 2015;12(3):415–440.
- SANDOZ. Leuprolide Acetate Injection [Internet]. p. 4. cited 2021 Dec 7]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/074728s011lbl.pdf
- ELIGARD® (leuprolide acetate for injectable suspension) | tolmar [Internet]. cited 2021 Dec 7]. Available from: https://www.tolmar.com/products/eligard
- Genovese S, Mannucci E, Ceriello A. A review of the long-term efficacy, tolerability, and safety of exenatide once weekly for type 2 diabetes. Adv Ther. 2017;34(8):1791–1814.
- Li T, Chandrashekar A, Beig A, et al. Characterization of attributes and in vitro performance of exenatide-loaded PLGA long-acting release microspheres. Eur J Pharm Biopharm. 2021;158:401–409.
- Blasi P. Poly(lactic acid)/poly(lactic-co-glycolic acid)-based microparticles: an overview. J Pharm Investig. 2019;49(4):337–346.
- Zhong H, Chan G, Hu Y, et al. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics. 2018;10(4):263.
- Moncalvo F, Martinez Espinoza MI, Cellesi F. Nanosized delivery systems for therapeutic proteins: clinically validated technologies and advanced development strategies. Front Bioeng Biotechnol. 2020;8:89.
- Lee WY, Asadujjaman M, Jee J-P. Long acting injectable formulations: the state of the arts and challenges of poly(lactic-co-glycolic acid) microsphere, hydrogel, organogel and liquid crystal. J Pharm Investig. 2019;49(4):459–476.
- Almeida M, Magalhães M, Veiga F, et al. Poloxamers, poloxamines and polymeric micelles: definition, structure and therapeutic applications in cancer. J Polym Res. 2018;25(1):31.
- Lupron Depot (leuprolide acetate for depot suspension) [Internet]. cited 2021 Nov 10]. Available from: https://www.lupron.com
- SANDOSTATIN® LAR Depot (octreotide acetate) for acromegaly and carcinoid syndrome [Internet]. cited 2021 Nov 10]. Available from: https://www.us.sandostatin.com
- Kempe S, Mäder K. In situ forming implants — an attractive formulation principle for parenteral depot formulations. J Control Release. 2012;161(2):668–679.
- Kanwar N, Sinha VR. In situ forming depot as sustained-release drug delivery systems. Crit Rev Ther Drug Carr Syst. 2019;36(2):93–136.
- About Zoladex® | zoladex® (goserelin acetate implant) [Internet]. cited 2021 Nov 15]. Available from: https://www.zoladex.com/what-is-zoladex
- [Internet]. cited 2021 Nov 5]. Available from: https://scenesse.com
- Mezzenga R, Seddon JM, Drummond CJ, et al. Nature‐inspired design and application of lipidic lyotropic liquid crystals. Adv Mater. 2019;31(35):1900818.
- Pipeline | Camurus [Internet]. cited 2021 Nov 8]. Available from: https://www.camurus.com/pipeline
- Forsback A-P, Noppari P, Viljanen J, et al. Sustained In-Vivo release of triptorelin acetate from a biodegradable silica depot: comparison to pamorelin® LA. Nanomaterials. 2021;11(6):1578.
- TNF-Bound colloidal gold in treating patients with advanced solid tumors - ClinicalTrials.gov [Internet]. cited 2021 Nov 9]. Available from: https://clinicaltrials.gov/ct2/show/NCT00356980.
- Pasut G. Grand challenges in nano-based drug delivery. Front Med Technol. 2019;1:1.
- Yu M, Wu J, Shi J, et al. Nanotechnology for protein delivery: overview and perspectives. J Control Release. 2016;240:24–37.
- Freitas S, Merkle HP, Gander B. Microencapsulation by solvent extraction/evaporation: reviewing the state of the art of microsphere preparation process technology. J Control Release. 2005;102(2):313–332.
- Sawant A, Kamath S, Kg H, et al. Solid-in-oil-in-water emulsion: an innovative paradigm to improve drug stability and biological activity. AAPS PharmSciTech. 2021;22(5):199.
- Butreddy A, Gaddam RP, Kommineni N, et al. PLGA/PLA-based long-acting injectable depot microspheres in clinical use: production and characterization overview for protein/peptide delivery. Int J Mol Sci. 2021;22(16):8884.
- Cai Y, Wei L, and Ma L, et al. Long-acting preparations of exenatide. Drug Des Devel Ther. 2013;7:963–970.
- Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int J Pharm. 2008;364(2):298–327.
- Stanković M, Frijlink HW, Hinrichs WLJ. Polymeric formulations for drug release prepared by hot melt extrusion: application and characterization. Drug Discov Today. 2015;20(7):812–823.
- Schwendeman SP, Shah RB, Bailey BA, et al. Injectable controlled release depots for large molecules. J Control Release. 2014;190:240–253.
- Zhou J, Walker J, Ackermann R, et al. Effect of manufacturing variables and raw materials on the composition-equivalent PLGA microspheres for 1-Month controlled release of leuprolide. Mol Pharm. 2020;17(5):1502–1515.
- Shi N-Q, Zhou J, Walker J, et al. Microencapsulation of luteinizing hormone-releasing hormone agonist in poly (lactic-co-glycolic acid) microspheres by spray-drying. J Control Release. 2020;321:756–772.
- Ogawa Y, Yamamoto M, Okada H, et al. A new technique to efficiently entrap leuprolide acetate into microcapsules of polylactic acid or copoly(Lactic/Glycolic) acid. Chem Pharm Bull (Tokyo). 1988;36(3):1095–1103.
- Ogawa Y, Yamamoto M, Takada S, et al. Controlled-Release of leuprolide acetate from polylactic acid or copoly(Lactic/Glycolic) acid microcapsules: influence of molecular weight and copolymer ratio of polymer. Chem Pharm Bull (Tokyo). 1988;36(4):1502–1507.
- Ogawa Y, Okada H, Yamamoto Y, et al. In vivo release profiles of leuprolide acetate from microcapsules prepared with polylactic acids or copoly(Lactic/Glycolic) acids and in vivo degradation of these polymers. Chem Pharm Bull (Tokyo). 1988;36(7):2576–2581.
- Beig A, Feng L, Walker J, et al. Physical–chemical characterization of octreotide encapsulated in commercial glucose-star PLGA microspheres. Mol Pharm. 2020;17(11):4141–4151.
- Zhou J, Hirota K, Ackermann R, et al. Reverse engineering the 1-Month Lupron Depot®. AAPS J. 2018;20(6):105.
- Ochi M, Wan B, Bao Q, et al. Influence of PLGA molecular weight distribution on leuprolide release from microspheres. Int J Pharm. 2021;599:120450.
- Wan B, Andhariya JV, Bao Q, et al. Effect of polymer source on in vitro drug release from PLGA microspheres. Int J Pharm. 2021;607:120907.
- Hadar J, Skidmore S, Garner J, et al. Characterization of branched poly(lactide-co-glycolide) polymers used in injectable, long-acting formulations. J Control Release. 2019;304:75–89.
- Na DH, Youn YS, Lee SD, et al. Monitoring of peptide acylation inside degrading PLGA microspheres by capillary electrophoresis and MALDI-TOF mass spectrometry. J Control Release. 2003;92(3):291–299.
- Shirangi M, Hennink WE, Somsen GW, et al. Identification and assessment of octreotide acylation in polyester microspheres by LC–MS/MS. Pharm Res. 2015;32(9):3044–3054.
- Molinier C, Picot-Groz M, Malval O, et al. Impact of octreotide counterion nature on the long-term stability and release kinetics from an in situ forming depot technology. J Control Release. 2021;336:457–468.
- Zhang Y, Sophocleous AM, Schwendeman SP. Inhibition of peptide acylation in PLGA microspheres with water-soluble divalent cationic salts. Pharm Res. 2009;26(8):1986–1994.
- Sophocleous AM, Zhang Y, Schwendeman SP. A new class of inhibitors of peptide sorption and acylation in PLGA. J Control Release. 2009;137(3):179–184.
- O’Brien MN, Jiang W, Wang Y, et al. Challenges and opportunities in the development of complex generic long-acting injectable drug products. J Control Release. 2021;336:144–158.
- Hua Y, Su Y, Zhang H, et al. Poly(lactic-co-glycolic acid) microsphere production based on quality by design: a review. Drug Deliv. 2021;28(1):1342–1355.
- Carrascosa C, Espejo L, Torrado S, et al. Effect of c-Sterilization Process on PLGA Microspheres Loaded with Insulin-Like Growth Factor - I (IGF-I). J Biomater Appl. 2003;18(2):95–108.
- Fineman M, Flanagan S, Taylor K, et al. Pharmacokinetics and pharmacodynamics of exenatide extended-release after single and multiple dosing. Clin Pharmacokinet. 2011;50(1):65–74.
- Nauck MA, Meier JJ. MANAGEMENT OF ENDOCRINE DISEASE: are all GLP-1 agonists equal in the treatment of type 2 diabetes? Eur J Endocrinol. 2019;181(6):R211–R234.
- Buss N, Ryan P, Baughman T, et al. Nonclinical safety and pharmacokinetics of Miglyol 812: a medium chain triglyceride in exenatide once weekly suspension: PK and safety of MCTs. J Appl Toxicol. 2018;38(10):1293–1301.
- Stewart S, Domínguez-Robles J, Donnelly R, et al. Implantable polymeric drug delivery devices: classification, manufacture, materials, and clinical applications. Polymers. 2018;10(12):1379.
- Sequeira JAD, Santos AC, Serra J, et al. Poly(lactic- co -glycolic acid) (PLGA) matrix implants. Nanostructures Eng Cells Tissues Organs [Internet]. Elsevier; 2018 [cited 2021 Nov 29]. p. 375–402. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128136652000107
- Jain A, Kunduru KR, Basu A, et al. Injectable formulations of poly(lactic acid) and its copolymers in clinical use. Adv Drug Deliv Rev. 2016;107:213–227.
- Kim ES, Garnock-Jones KP. Afamelanotide: a review in erythropoietic protoporphyria. Am J Clin Dermatol. 2016;17(2):179–185.
- Montgomery BSI, Borwell JP, Higgins DM. Does needle size matter? Patient experience of luteinising hormone-releasing hormone analogue injection. Prostate Cancer Prostatic Dis. 2005;8(1):66–68.
- Southard G, Dunn R, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL®technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998;7(9):1483–1491.
- Shore N, Mincik I, DeGuenther M, et al. A phase 3, open-label, multicenter study of a 6-month pre-mixed depot formulation of leuprolide mesylate in advanced prostate cancer patients. World J Urol. 2020;38(1):111–119.
- Zhou H, Hernandez C, Goss M, et al. Biomedical imaging in implantable drug delivery systems. Curr Drug Targets. 2015;16(6):672–682.
- Roberge C, Cros J-M, Serindoux J, et al. BEPO®: bioresorbable diblock mPEG-PDLLA and triblock PDLLA-PEG-PDLLA based in situ forming depots with flexible drug delivery kinetics modulation. J Control Release. 2020;319:416–427.
- Saltzstein D, Shore ND, Moul JW, et al. Pharmacokinetic and pharmacodynamic comparison of subcutaneous versus intramuscular leuprolide acetate formulations in male subjects. Ther Adv Urol. 2018;10(2):43–50.
- Mostafa NM, Chwalisz K, Larsen L, et al. Evaluation of the pharmacokinetics and pharmacodynamics of two leuprolide acetate 45 mg 6-month depot formulations in patients with prostate cancer: clinical pharmacology in drug development. Clin Pharmacol Drug Dev. 2014;3(4):270–275.
- Park K, Skidmore S, Hadar J, et al. Injectable, long-acting PLGA formulations: analyzing PLGA and understanding microparticle formation. J Control Release. 2019;304:125–134.
- Klein KO, Freire A, Gryngarten MG, et al. Phase 3 trial of a small-volume subcutaneous 6-Month duration leuprolide acetate treatment for central precocious puberty. J Clin Endocrinol Metab. 2020;105(10):e3660–e3671.
- Tiberg F, Roberts J, Cervin C, et al. Octreotide s.c. depot provides sustained octreotide bioavailability and similar IGF‐1 suppression to octreotide LAR in healthy volunteers. Br J Clin Pharmacol. 2015;80(3):460–472.
- Richard J, Prang N. The formulation and immunogenicity of therapeutic proteins: product quality is a key factor. Thomson Reuters. 2010;13:550–558.
- Lee PW, Pokorski JK. PLGA devices: production and applications for sustained protein delivery. 2019;38.
- Estey T, Kang J, Schwendeman SP, et al. BSA degradation under acidic conditions: a model for protein instability during release from PLGA delivery systems. J Pharm Sci. 2006;95(7):1626–1639.
- Mohammadi-Samani S, Taghipour B. PLGA micro and nanoparticles in delivery of peptides and proteins; problems and approaches. Pharm Dev Technol. 2015;20(4):385–393.
- Wang W, Nema S, Teagarden D. Protein aggregation—Pathways and influencing factors. Int J Pharm. 2010;390(2):89–99.
- Wang W, Ohtake S. Science and art of protein formulation development. Int J Pharm. 2019;568:118505.
- Sah H. Stabilization of proteins against methylene chloride/water interface-induced denaturation and aggregation. J Control Release. 1999;58(2):143–151.
- Kamerzell TJ, Esfandiary R, Joshi SB, et al. Protein–excipient interactions: mechanisms and biophysical characterization applied to protein formulation development. Adv Drug Deliv Rev. 2011;63(13):1118–1159.
- Moreno MR, Tabitha TS, Nirmal J, et al. Study of stability and biophysical characterization of ranibizumab and aflibercept. Eur J Pharm Biopharm. 2016;108:156–167.
- Lee PW, Pokorski JK. Poly(lactic‐co‐glycolic acid) devices: production and applications for sustained protein delivery. WIREs Nanomed Nanobiotechnol. 2018;10(5). 10.1002/wnan.1516
- Arakawa T, Kita Y, Timasheff SN. Protein precipitation and denaturation by dimethyl sulfoxide. Biophys Chem. 2007;131(1–3):62–70.
- Leconet W, Liu H, Guo M, et al. Anti-PSMA/CD3 bispecific antibody delivery and antitumor activity using a polymeric depot formulation. Mol Cancer Ther. 2018;17(9):1927–1940.
- Chang DP, Garripelli VK, Rea J, et al. Investigation of fragment antibody stability and its release mechanism from poly(Lactide-co-Glycolide)-triacetin depots for sustained-release applications. J Pharm Sci. 2015;104(10):3404–3417.
- Nejadnik MR, Randolph TW, Volkin DB, et al. Postproduction handling and administration of protein pharmaceuticals and potential instability issues. J Pharm Sci. 2018;107(8):2013–2019.
- Bee JS, Stevenson JL, Mehta B, et al. Response of a concentrated monoclonal antibody formulation to high shear. Biotechnol Bioeng. 2009;103(5):936–943.
- Zhang C, Yang L, Wan F, et al. Quality by design thinking in the development of long-acting injectable PLGA/PLA-based microspheres for peptide and protein drug delivery. Int J Pharm. 2020;585:119441.
- Le M-Q, Violet F, Paniagua C, et al. Penta-block copolymer microspheres: impact of polymer characteristics and process parameters on protein release. Int J Pharm. 2018;535(1–2):428–437.
- Zhu G, Schwendeman SP Stabilization of Proteins Encapsulated in Cylindrical Poly(lactide-co-glycolide) Implants: mechanism of Stabilization by Basic Additives.:7.
- Aubert-Pouëssel A, Bibby DC, Venier-Julienne M-C, et al. A novel in vitro delivery system for assessing the biological integrity of protein upon release from PLGA microspheres. Pharm Res. 2002;19(7):1046–1051.
- Duralliu A, Matejtschuk P, Stickings P, et al. The influence of moisture content and temperature on the long-term storage stability of freeze-dried high concentration immunoglobulin G (IgG). Pharmaceutics. 2020;12(4):303.
- Ohori R, Akita T, Yamashita C. Effect of temperature ramp rate during the primary drying process on the properties of amorphous-based lyophilized cake, Part 2: successful lyophilization by adopting a fast ramp rate during primary drying in protein formulations. Eur J Pharm Biopharm. 2018;130:83–95.
- Ferber S, Behrens AM, McHugh KJ, et al. Evaporative cooling hydrogel packaging for storing biologics outside of the Cold Chain. Adv Healthc Mater. 2018;7(14):1800220.
- Brader ML, Estey T, Bai S, et al. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12(4):1005–1017.
- Bommana R, Chai Q, Schöneich C, et al. Understanding the increased aggregation propensity of a light-exposed IgG1 monoclonal antibody using hydrogen exchange mass spectrometry, biophysical characterization, and structural analysis. J Pharm Sci. 2018;107(6):1498–1511.
- Wang W, Ignatius AA, Thakkar SV. Impact of residual impurities and contaminants on protein stability. J Pharm Sci. 2014;103(5):1315–1330.
- Qi F, Wu J, Li H, et al. Recent research and development of PLGA/PLA microspheres/nanoparticles: a review in scientific and industrial aspects. Front Chem Sci Eng. 2019;13(1):14–27.
- Vehring R, Snyder H, and Lechuga‐Ballesteros D. Spray Drying. In: Ohtake S, Izutsu K, Lechuga‐Ballesteros D, editors. Drying Technologies for Biotechnoly and Pharmaceutical Applications. 1st ed. NJ, USA: Wiley; 2020. p. 179–216.
- Pinto JT, Faulhammer E, Dieplinger J, et al. Progress in spray-drying of protein pharmaceuticals: literature analysis of trends in formulation and process attributes. Dry Technol. 2021;39(11):1415–1446.
- Pham NB, Meng WS. Protein aggregation and immunogenicity of biotherapeutics. Int J Pharm. 2020;585:119523.
- Kozma GT, Shimizu T, Ishida T, et al. Anti-PEG antibodies: properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv Drug Deliv Rev. 2020;154–155:163–175.
- Immunogenicity Assessment for Therapeutic Protein Products | FDA [Internet]. cited 2021 Dec 17]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-assessment-therapeutic-protein-products
- Yu L, Sun Q, Hui Y, et al. Microfluidic formation of core-shell alginate microparticles for protein encapsulation and controlled release. J Colloid Interface Sci. 2019;539:497–503.
- Kirchhof S, Gregoritza M, Messmann V, et al. Diels–Alder hydrogels with enhanced stability: first step toward controlled release of bevacizumab. Eur J Pharm Biopharm. 2015;96:217–225.
- Fayd’herbe, De Maudave A, Leconet W, Toupet K, et al. Intra-articular delivery of full-length antibodies through the use of an in situ forming depot. J Control Release. 2022;341:578–590.
- Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res. 2011;28(10):2379–2385.
- Rombach-Riegraf V, Karle AC, and Wolf B, et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. In: PLoS ONE. Vol. 9. 2014. pp. e86322.
- Joubert MK, Deshpande M, and Yang J, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. In: PLOS ONE. Vol. 11. 2016. e0159328.
- Morgan H, Tseng S-Y, Gallais Y, et al. Evaluation of in vitro assays to assess the modulation of dendritic cells functions by therapeutic antibodies and aggregates. Front Immunol. 2019;10:601.
- Lu Y, Kim S, Park K. In vitro–in vivo correlation: perspectives on model development. Int J Pharm. 2011;418(1):142–148.
- Olthof MGL, Tryfonidou MA, Dadsetan M, et al. In vitro and in vivo correlation of bone morphogenetic protein-2 release profiles from complex delivery vehicles. Tissue Eng Part C Methods. 2018;24(7):379–390.
- Jain P, Pawar RS, Pandey RS, et al. In-vitro in-vivo correlation (IVIVC) in nanomedicine: is protein Corona the missing link? Biotechnol Adv. 2017;35(7):889–904.
- Zolnik BS, Burgess DJ. In vitro–in vivo correlation on parenteral dosage forms. In Krishna R, Yu L, editors. Biopharm appl drug dev Internet]. Boston (MA): Springer US; 2008, 336–358. [cited 2021 Nov 23]. Available from: http://link.springer.com/10.1007/978-0-387-72379-2_11
- Lal RA, Hoffman AR. Long-acting growth hormone preparations in the treatment of children. 2019;9.
- Teekamp N, Van Dijk F, Broesder A, et al. Polymeric microspheres for the sustained release of a protein-based drug carrier targeting the PDGFβ-receptor in the fibrotic kidney. Int J Pharm. 2017;534(1–2):229–236.
- Polymer technologies - Innocore Pharma [Internet]. cited 2021 Nov 29]. Available from: https://www.innocorepharma.com/nl/Technologies/Polymer%20technologies/SynBiosys%C2%AE
- Stanković M, Tomar J, Hiemstra C, et al. Tailored protein release from biodegradable poly(ε-caprolactone-PEG)-b-poly(ε-caprolactone) multiblock-copolymer implants. Eur J Pharm Biopharm. 2014;87(2):329–337.
- Tyagi P, Koskinen M, Mikkola J, et al. Silica microparticles for sustained zero-order release of an anti-CD40L antibody. Drug Deliv Transl Res. 2018;8(2):368–374.
- Libutti SK, Paciotti GF, Byrnes AA, et al. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res. 2010;16(24):6139–6149.
- [Internet]. cited 2022 Apr 12]. Available from: https://www.cytimmune.com/pipelilne
- Chan Y-P, Meyrueix R, Kravtzoff R, et al. Review on Medusa ®:a polymer-based sustained release technology for protein and peptide drugs. Expert Opin Drug Deliv. 2007;4(4):441–451.
- OctoPlus. PolyActiveTM A biodegradable polymer-based drug delivery system [Internet]. 2006 cited 2021 Dec 2]. Available from: http://www.afinitica.com/arnews/sites/default/files/techdocs/PolyActive_Nov20061%5B1%5D.pdf
- Biolex Therapeutics, Inc. An Open-Label, 3-Panel, Dose-Escalation Study to Assess the Safety and Tolerability, Pharmacokinetics, and Viral Kinetics of Two Doses of LocteronTM (Poly ActiveTM - Interferon Alpha 2b) Given Every 2 Weeks for 4-12 Weeks in Comparison With PEG-Intron Given Weekly for 4-12 Weeks in Patients With Chronic Hepatitis C [Internet]. clinicaltrials.gov; 2012 [cited 2021 Dec 1]. Report No.: NCT00593151. Available from: https://clinicaltrials.gov/ct2/show/NCT00593151.
- Biolex therapeutics researchers present Locteron® U.S. Phase 2a Hepatitis C Trial Results at EASL Conference | proteins and Peptides | news Channels [Internet]. cited 2021 Dec 1]. Available from: https://pipelinereview.com/index.php/2009042426544/Proteins-and-Peptides/Biolex-Therapeutics-Researchers-Present-Locteron-U.S.-Phase-2a-Hepatitis-C-Trial-Results-at-EASL-Conference.html
- Ramos TI, Villacis-Aguirre CA, Santiago Vispo N, et al. Forms and methods for interferon’s encapsulation. Pharmaceutics. 2021;13(10):1533.
- Technology – bioChaperone® – ADOCIA a biotechnology company dedicated to protein delivery [Internet]. cited 2021 Nov 29]. Available from: https://www.adocia.com/technology/biochaperone-technology-2
- Products – bioChaperone® ultra-rapid insulin – ADOCIA a biotechnology company dedicated to protein delivery [Internet]. cited 2021 Dec 1]. Available from: https://www.adocia.com/products/biochaperone-ultra-fast-analog-insulin
- Adocia ends BioChaperone PDGF development after phase III Failure [Internet]. cited 2021 Dec 1]. Available from: https://www.genengnews.com/news/adocia-ends-biochaperone-pdgf-development-after-phase-iii-failure
- Serwer L, Hashizume R, and Ozawa T, et al. Systemic and local drug delivery for treating diseases of the central nervous system in rodent models. J Vis Exp. 2010;e1992.
- Zhang Y, Jordan JM. Epidemiology of osteoarthritis. Clin Geriatr Med. 2010;26(3):355–369.
- Evans CH, Kraus VB, Setton LA. Progress in intra-articular therapy. Nat Rev Rheumatol. 2014;10(1):11–22.
- Fayd’herbe De Maudave A, Leconet W, Toupet K, et al. Intra-articular delivery of full-length antibodies through the use of an in situ forming depot. J Control Release. 2022;341:578–590
- Shi WJ, Tjoumakaris FP, Lendner M, et al. Biologic injections for osteoarthritis and articular cartilage damage: can we modify disease? Phys Sportsmed. 2017;45(3):203–223.
- Intra-tumoral ipilimumab plus intravenous nivolumab following the resection of recurrent glioblastoma - ClinicalTrials.gov [Internet]. cited 2021 Apr 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT03233152
- Intratumoral Tilsotolimod, a TLR-9 agonist, together with intratumoral ipilimumab and intravenous nivolumab in patients with advanced cancers - ClinicalTrials.gov [Internet]. cited 2021 Apr 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT04270864
- A trial evaluating the safety & efficacy of intra-tumoral ipilimumab in combination with intra-venous nivolumab in patients with metastatic melanoma - clinicalTrials.gov [Internet]. cited 2021 Apr 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT02857569
- Heldin C-H, Rubin K, Pietras K, et al. High interstitial fluid pressure — an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813.
- Nam J, Son S, Park KS, et al. Cancer nanomedicine for combination cancer immunotherapy. Nat Rev Mater. 2019;4(6):398–414.
- Bhatia S, Miller NJ, Lu H, et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with Merkel cell carcinoma. Clin Cancer Res. 2019;25(4):1185–1195.
- Marabelle A, Andtbacka R, Harrington K, et al. Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann Oncol. 2018;29(11):2163–2174.
- Muñoz NM, Williams M, Dixon K, et al. Influence of injection technique, drug formulation and tumor microenvironment on intratumoral immunotherapy delivery and efficacy. J Immunother Cancer. 2021;9(2):e001800.
- Peyman GA, Lad EM, Moshfeghi DM. Intravitreal injection of therapeutic agents. Retina. 2009;29(7):875–912.
- Otero-Espinar FJ, Fernández-Ferreiro A, González-Barcia M, et al. Stimuli sensitive ocular drug delivery systems. Drug target stimuli sensitive drug deliv syst [Internet]. Elsevier; 2018 [cited 2021 Nov 26]. p. 211–270. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128136898000069.
- Chandrasekaran PR, Madanagopalan VG. KSI-301: antibody biopolymer conjugate in retinal disorders. Ther Adv Ophthalmol. 2021;13:251584142110277.
- Brown TD, Whitehead KA, Mitragotri S. Materials for oral delivery of proteins and peptides. Nat Rev Mater. 2020;5:127–148.
- Brayden DJ, Hill TA, Fairlie DP, et al. Systemic delivery of peptides by the oral route: formulation and medicinal chemistry approaches. Adv Drug Deliv Rev. 2020;157:2–36.
- Karsdal MA, Byrjalsen I, Riis BJ, et al. Optimizing bioavailability of oral administration of small peptides through pharmacokinetic and pharmacodynamic parameters: the effect of water and timing of meal intake on oral delivery of salmon calcitonin. BMC Clin Pharmacol. 2008;8(1):5.
- Richard J. Formulation Strategies based on Supercritical Fluid Technology for the Delivery of Biopharmaceuticals. Chem Today. 2008;26:48–51.
- Lewis AL, Richard J. Challenges in the delivery of peptide drugs: an industry perspective. Ther Deliv. 2015;6(2):149–163.
- Cherif-Cheikh R, Delgado De Sousa A-P, Lacombe F, et al. Sustained release formulations comprising very low molecular weight polymers.
- Richard J. Challenges in oral peptide delivery: lessons learnt from the clinic and future prospects. Ther Deliv. 2017;8(8):663–684.
- Jamaledin R, Di Natale C, Onesto V, et al. Progress in microneedle-mediated protein delivery. J Clin Med. 2020;9(2):542.
- Vora LK, Moffatt K, Tekko IA, et al. Microneedle array systems for long-acting drug delivery. Eur J Pharm Biopharm. 2021;159:44–76.
- Daddona PE, Matriano James A, Mandema J, et al. Parathyroid hormone (1-34)-coated microneedle patch system: clinical pharmacokinetics and pharmacodynamics for treatment of osteoporosis. Pharm Res. 2011;28(1):159–165.
- Kochba E, Levin Y, Raz I, et al. Improved insulin pharmacokinetics using a novel microneedle device for intradermal delivery in patients with type 2 diabetes. Diabetes Technol Ther. 2016;18(9):525–531.
- Jacobse J, Voorde W, Tandon A, et al. Comprehensive evaluation of microneedle-based intradermal Adalimumab delivery vs. subcutaneous administration: results of a randomized controlled clinical trial. Br J Clin Pharmacol. 2021;87(8):3162–3176.